


INTRODUCTION
Genitourinary tract infections due to Neisseria gonorrhoeae is a major cause of morbidity in sexually active individuals [1, Reference Mahony2]. In an effort to prevent spread of this disease, increased attention is being focused on early diagnosis and treatment of symptomatic as well as asymptomatic individuals, which are not being adequately addressed by the conventional diagnostic methods, i.e. microscopy and culture [Reference Hammerschlag3]. Hence, various nucleic acid amplification tests (NAATs), predominantly polymerase chain reactions (PCRs) targeting different genes are being developed and evaluated by laboratories worldwide and include opa, 16S rRNA, porA pseudogene, cppB gene, CMT gene and the pil gene. A huge variation in sensitivity and specificity of these gene targets has been reported mainly depending on the primers and probes used and the population where they were evaluated [Reference Smith, Tapsall and Lum4]. Therefore, any N. gonorrhoeae NAAT must be validated in the prospective patient population prior to being introduced as a routine diagnostic test. Another major drawback of NAATs with respect to detection of N. gonorrhoeae is the cross-reactivity seen with non-N. gonorrhoeae Neisseria species (non-NgNS) [Reference Katz5, Reference Tabrizi6]. Therefore, a single PCR targeting one gene cannot be relied upon for definitive diagnosis of gonorrhoea in any patient population.
The Centers for Disease Control and Prevention (CDC), Australian Public Health Laboratory Network (PHLN) and the UK's Health Protection Agency (HPA) guidelines, recommend the use of two PCRs targeting different genes known to have a discriminatory capacity. The PCR screening assay should be highly sensitive while the supplementary assay should be highly specific and have a sensitivity comparable to the screening assay [Reference Smith, Tapsall and Lum4, Reference Johnson7, 8]. Indeed, based on current US recommendations for Chlamydia and gonorrhoea testing and as reinforced by the PHLN, the acceptable performance criteria for test combinations requires that the reported result should have a positive predictive value (PPV) of at least 90% in the population being tested [Reference Johnson7, 8]. Further, as recommended by the PHLN guidelines an on-going assessment of various gene assays is required [Reference Smith, Tapsall and Lum4]. The objective of the present study was to develop and evaluate the performance of an in-house PCR assay based on the opa gene compared to the porA pseudogene and 16S rRNA gene-based assays. In accordance with our earlier report, the 16S rRNA assay with its high sensitivity has been used as the screening assay and the highly specific porA pseudogene-based assay as the supplementary assay for detection of positives [Reference Verma9].
METHODS
Bacterial isolates and clinical samples
The panel of strains used for evaluation comprised N. gonorrhoeae (ATCC 49226, WHO B, WHO C, WHO F, WHO G, WHO K, WHO L, WHO M, WHO N, WHO O, WHO P; quality control strains received since 2005 as a part of the External Quality Assurance Scheme conducted by the Regional Reference Laboratory, Safdarjang Hospital, New Delhi (n=26); patient isolates (n=14); Chlamydia trachomatis, Mycoplasma genitalium, Mycoplasma hominis (NCTC 10111), Ureaplasma urealyticum (NCTC 10177), Streptococcus pyogenes (ATCC 19615), group B streptococci (clinical isolate), Candida spp., Klebsiella pneumoniae (ATCC 700603), Pseudomonas aeruginosa (ATCC 27853), Staphylococcus aureus (ATCC 25923), Escherichia coli (ATCC 25922), Enterococcus faecalis (ATCC 51299), N. meningitidis [ATCC 13077 serogroup A (n=1), clinical isolate (n=3)] and N. sicca (ATCC 29193 (n=1), clinical isolates (n=8)].
Sample sizes of the populations to be screened were determined on the basis of prevalence data. Considering the positivity rate in males as 40%, to estimate this prevalence with absolute precision of ±10 with 95% confidence level, required 100 males and similarly for females with a positivity rate of 7%, to estimate this prevalence with absolute precision of ±3 with 95% confidence level, the required number was calculated as 300. Four hundred patients (100 males, 300 females) aged 15–45 years with symptoms suggestive of gonococcal infections visiting the STD clinics, Dermatology Outpatient Department of the All India Institute of Medical Sciences (AIIMS) and the Regional STD, Teaching Training and Research Centre, Safdarjung Hospital, New Delhi, India were included in the study. The inclusion criteria for patients were urethral, cervical/vaginal discharge or lower abdominal pain. In addition, history of symptomatic partner or positive risk factors or partner with risk factors was taken into account for inclusion criteria. Urethral/endocervical swabs were collected in triplicate by standard protocol by a trained physician [10]: one swab was used to prepare a smear for microscopic examination, a second for culture and the third swab for DNA extraction for PCR.
The study was approved by the Ethics Committee, AIIMS, New Delhi, India.
Sample processing
The smear was Gram stained by standard method and swabs were cultured on saponin-lysed blood agar plus VCNT(A) inhibitors and chocolate agar. Suspected colonies were confirmed by oxidase test, superoxol and rapid carbohydrate utilization tests [10]. DNA was extracted from bacterial cultures and the third swab sample using QIAamp DNA mini kit (Qiagen Sciences Inc., USA) according to the manufacturer's instructions.
opa gene-based PCR
For the opa gene-based PCR, primers were designed using the available software ClustalX and Primer3 from the gene sequences available in the GenBank database. The selected oligonucleotide sequence was then analysed for non-specific binding using BLAST. The forward primer was 5′-CGG TGC TTC ATC ACC TTA G-3′ and the reverse primer was 5′-GGA TTC ATT TTC GGC TCC TT-3′. Sequences were validated by GenBank (GenBank accession no. PUID 9716120 SNUM 2706 Ng_opa). The optimum annealing temperature for the PCR was determined from a gradient range of 50–60°C and the optimal MgCl2 concentration from a range of MgCl2 (1·5–2·5 mm). PCR was also performed with different concentrations of Taq polymerase (0·75–2·5 U) and forward and reverse primers (4–6 pmol). A 25-μl reaction contained a final concentration of reaction buffer (10 mm KCl, 10 mm HCl), 1·5 mM MgCl2, Taq polymerase (1·5 U), 200 μm dNTP mix and 5 pmol primers. PCR conditions were denaturation at 94°C for 45 s, annealing at 56°C for 45 s and extension at 72°C for 45 s for a total of 30 cycles yielding a 188-bp product from positive samples.
porA pseudogene PCR
For porA pseudogene PCR primers 5′-CCG GAA CTG GTT TCA TCT GAT T-3′ and 5′-GTT TCA GCG GCA GCA TTC A-3′ were used [Reference Hjelmevoll11]. The reaction mix (25 μl) was as above for the opa gene PCR and cycling conditions were denaturation at 95°C for 45 s, annealing at 60°C for 45 s and extension at 72°C for 45 s for a total of 30 cycles yielding a 102-bp product from positive samples.
16S rRNA PCR
The forward and reverse primers were as described previously [Reference Farrell12] utilizing the same reaction mix with denaturation at 95°C for 45 s, annealing at 68°C for 1 min and extension at 72°C for 1 min for a total of 40 cycles to yield a 413-bp product.
All PCR assays were controlled for sample inhibition by the amplification of the β-globin gene as previously described [Reference Verma9].
Sensitivity and specificity
Tenfold dilutions were prepared from DNA extracted from a standard strain of N. gonorrhoeae to determine the detection limit of each of the PCR assays. Specificity was determined by reactivity of each PCR with non-NgNS and other control species in the strain panel.
Statistical analysis
True positives were defined as samples positive by culture and/or both the 16S rRNA and porA pseudogene-based assays [Reference Verma9]. The 95% confidence interval was calculated according to the efficient score method (corrected for continuity) using the online tool available at http://faculty.vassar.edu/lowry/VassarStats.html.
RESULTS
Microscopy and culture
Out of 400 patients (100 males, 300 females) 46 (44 males, 2 females) were positive by microscopy and 44 (42 males, 2 females) by culture.
Performance characteristics of PCR assays
Specificity tests confirmed that no species other than N. gonorrhoeae reacted in the opa and porA pseudogene PCR assays but cross-reactivity with N. sicca was found with the 16S rRNA assay. The limit of detection of the opa and 16S rRNA assays was determined as 0·4 pg N. gonorrhoeae DNA but the porA pseudogene-based PCR was tenfold less sensitive (4 pg DNA).
Performance of PCR assays in patient samples
The 16S rRNA assay was positive in 82, opa in 67 and porA assay in 60 patients (Table 1). Sixty patient samples were classified as ‘true positives’ on the basis of their positivity in both the porA and 16S rRNA assays.
Table 1. Comparison of opa PCR with porA and 16S ribosomal assay

* These seven patients were also positive by the 16S rRNA PCR.
Of the 67 patients positive by opa assay, 60 were positive by both 16S and porA and seven only by 16S rRNA. The sensitivity, specificity, PPV and negative predictive value (NPV) of the assay based on the 60 confirmed positives was 100% (95% CI 92·5–100), 97·9% (95% CI 95·6–99·1), 89·5% (95% CI 79·1–95·3) and 100% (95% CI 98·6–100), respectively. Of the seven discrepant samples positive by opa assay which were positive in both the opa assay and the 16S rRNA assay but discrepant in the porA assay, opa amplicons for four samples could be sequenced; the other three had faint bands and hence could not be sequenced. The sequences gave a perfect match with the N. gonorrhoeae opacity gene sequence available in GenBank (three with gonococcal protein II opacity gene and one with opacity gene variant V28), confirming that the DNA being amplified in the patient samples was from N. gonorrhoeae (Fig. 1 a, b).

Fig. 1. (a) Sequence alignment of opa amplicons of three patients with protein II opacity gene of N. gonorrhoeae. (b) Sequence alignment of opa amplicon of one patient with opacity gene variant V28 of N. gonorrhoeae.
DISCUSSION
There has been a significant change in the approach to the diagnosis of gonorrhoea in recent years and NAATs are now routinely used for detection of N. gonorrhoeae DNA in clinical samples. However, to date no single gonococcal NAAT has proved to be both sensitive and specific across a broad range of patient populations perhaps due to the well developed capacity of the gonococcus for genetic variation and recombination [Reference Tabrizi6, Reference Whiley, Tapsall and Sloots13]. These characteristics coupled with the fastidious nature of the organism have created considerable difficulties for the laboratory diagnosis of gonorrhoea using conventional as well as molecular approaches.
In this study an in-house conventional PCR targeting the opa gene was standardized and its analytical and diagnostic performance was determined and compared against two published PCR assays [Reference Hjelmevoll11, Reference Farrell12]. The porA pseudogene and the opa-based PCR assays proved to be highly specific for gonococcal DNA whereas cross-reaction with N. sicca was evident with the 16S rRNA assay. The high specificity of the porA pseudogene-based assay was in accordance with other studies which have shown high conservation of this gene across N. gonorrhoeae subtypes [Reference Whiley, Tapsall and Sloots13, Reference Whiley14]. Similarly, cross-reactivity of 16S genes with commensal Neisseria spp. has been observed by others [Reference Boel15] and it is possible that different primer sequences to amplify this gene may improve specificity of the assay. The sensitivity of the porA pseudogene assay was tenfold less than the other assays perhaps owing to its presence as a single copy in the gonococcal genome. Additionally, the published primers used in this study were designed for real-time PCR in conjunction with probes which might account for the lower sensitivity also reported by other studies [Reference Mangold16, Reference Maze17]. Increasing the number of cycles, modification of reaction conditions may improve the sensitivity of this PCR.
The sequence homology of the seven discrepant results (16S rRNA+, opa+, porA−) with N. gonorrhoeae standard sequence suggests that these patient samples were indeed ‘true positives’ implying that the specificity and PPV of the assay was perhaps better than the projected figures. The discrepancy in the result may be due to the lower sensitivity of the porA pseudogene. The recent report of sequence variation in porA pseudogene further raises concern over the impact of sequence variation on the performance of gonococcal NAATs [Reference Whiley18].
The 16S rRNA PCR revealed 15 positive samples negative by both other assays. As the sensitivity of the opa assay is equivalent to the 16S rRNA assay it was considered unlikely that these samples were false negative by the opa PCR and hence they were not processed further. In fact the opa gene-based assay is reported to be five times more sensitive than the 16S rRNA assay presumably due to the higher copy number (n=11) of the former compared to the latter (n=4) in the N. gonorrhoeae genome [Reference Geraats-Peters19]. It is noteworthy that all of the discrepant 16S rRNA results were from female patients and this might have been due to cross-contamination of the assay with the diverse microbial flora of the female genital tract and the contribution of the high rate of horizontal genetic exchange within the Neisseria genus. Further, N. gonorrhoeae species comprises a broad range of subtypes which exhibit considerable genetic variation. The continually changing prevalence of these subtypes in a patient population can have a significant impact on the success of any N. gonorrhoeae NAAT [Reference Whiley, Tapsall and Sloots13]. Therefore, assays need to be routinely monitored in order to identify any new variation in the target sequence used.
The conservation and stability of target DNA also has a major impact on the sensitivity of the assay. Other authors have also reported the opa gene-based assay to be sensitive, specific and reliable for detection of N. gonorrhoeae in clinical samples and/or for confirmation of less specific tests [Reference Whiley, Tapsall and Sloots13, Reference Tabrizi20]. However, a recent study has described sequence variation in the multicopy opa gene leading to false-negative results [Reference Geraats-Peters19]. Although, alignment of the sequence amplified by the in-house opa PCR with the target sequences of the earlier studies [Reference Geraats-Peters19, Reference Tabrizi20] showed that the amplified product overlaps the target sequence in question (Fig. 2), the gene remains a highly stable target. By contrast with the earlier report by Geraats-Peters et al. [Reference Geraats-Peters19], the present study picked up seven extra positives by the opa-based assay, four of which could be sequenced and were confirmed as true positives by DNA sequencing. Therefore, we have reason to believe that as suggested by others, variation observed in the opa gene of Dutch gonococci may not yet be as widespread [Reference Goire21]. Unlike the real-time PCR formats used by others [Reference Geraats-Peters19, Reference Tabrizi20] our opa assay is a conventional PCR which after more extensive validation in different population groups would be suitable for laboratories which do not have access to real-time PCR machines. An investigation into the stability of the target sequence across a diverse range of gonococcal isolates from across the country may provide the necessary insight.
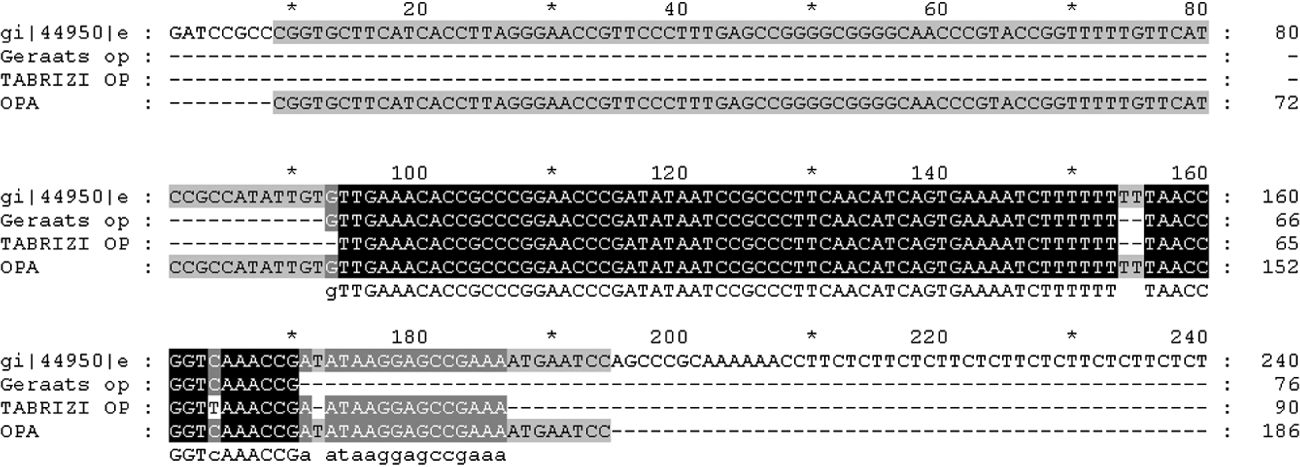
Fig. 2. Alignment of DNA sequence of opa amplicon with target sequences of Geraats-Peters et al. [Reference Geraats-Peters19] and Tabrizi et al. [Reference Tabrizi20] with respect to protein II (pFLOB1100) of N. gonorrhoeae.
Although not surprising, an interesting finding was the low clinical sensitivity of conventional methods for the detection of N. gonorrhoeae particularly in female patients (21 false negatives). Even in male patients where the performance characteristic of culture is known to be good, there were two false negatives. Therefore, where possible diagnostic laboratories should adopt molecular techniques in conjunction with standard culture to optimize the detection and hence control this bacterial sexually transmitted infection. However, a close watch on the antimicrobial resistance of N. gonorrhoeae is mandatory but this at present cannot be met by NAATs.
To conclude we have shown an in-house opa-based PCR assay to be highly sensitive and specific for detection of N. gonorrhoeae in clinical swab samples. This assay may prove to be a suitable supplementary test in our population along with 16S rRNA as a screening assay. Moreover, the use of NAATs provided enhanced diagnosis of gonorrhoea in female patients.
ACKNOWLEDGEMENTS
The work was supported by grant no. BT/PR7667/MED/14/1057/2006 from the Department of Biotechnology, Ministry of Science and Technology, Government of India. The authors thank the Council of Scientific and Industrial Research, India for financial assistance. We also thank Dr B. D. Malhotra, Scientist G, Dr G. Sumana, Scientist C and Renu Singh, Ph.D. student, BECPRL, National Physical Laboratory, New Delhi, India for their valuable inputs. We are grateful to Dr Aruna Mittal, Scientist F, National Institute of Pathology, ICMR, for providing us with C. trachomatis DNA. We are also grateful to Dr Benu Dhawan, Additional Professor, Department of Microbiology, AIIMS, for providing us DNA of Mycoplasma hominis (NCTC 10111), Mycoplasma genitalium and Ureaplasma urealyticum (NCTC 10177) and to Dr Immaculata Xess, Additional Professor, Department of Microbiology, AIIMS for providing DNA of Candida spp. We also thank Ms M. Kalaivani, Scientist, Department of Biostatistics, AIIMS, New Delhi for providing statistical analysis and Mr Rajinder Singh, Laboratory Technician, STD Laboratory, Department of Microbiology, AIIMS, for his invaluable assistance.
DECLARATION OF INTEREST
None.



