Alzheimer disease (AD) is characterised by a slowly progressive decline of memory and cognition. Alzheimer described the characteristic cortical senile plaques and neurofibrillary tangles in the brain of a 51-year-old woman with presenile dementia (Refs Reference Alzheimer1, Reference Alzheimer2). Because the presenile form, with onset before the age of 65, is identical to the most common form of senile dementia, today, the term AD is used for the designation of both presenile and senile cases (Refs Reference Katzman3, Reference Terry and Davies4).
Senile plaques were first described by Blocq and Marinesco (Ref. Reference Blocq and Marinesco5). Redlich (Ref. Reference Redlich6) first observed senile plaques in the brains of two patients with senile dementia. Recently, particularly from the use of the Gallyas silver technique (Ref. Reference Gallyas7), neuropil threads or curly fibres were recognised as further characteristic lesions of AD. Granulovacuolar degeneration is another typical alteration of neurons (Ref. Reference Simchowicz8). Other important features include neuronal and synaptic loss (Ref. Reference Terry9), Hirano bodies, reactive astrocytosis and microgliosis.
AD is a form of amyloidosis. The amyloid substance aggregated in the brain is a small, ~4 kDa amyloid beta peptide (Aβ). It is released by β- and γ-secretase cleavage from a larger 120 kDa transmembrane amyloid precursor protein (APP) (Refs Reference Kang10, Reference Selkoe11) and was purified and partially sequenced by Glenner and Wong (Ref. Reference Glenner and Wong12). APP exhibits features of a glycosylated cell-surface receptor and was shown to be a proteoglycan core protein (Ref. Reference Schubert13). APP is phylogenetically highly conserved and constitutively expressed by various cells other than neurons, including immune cells (Ref. Reference Bush, Beyreuther and Masters14). Human platelets, peripheral lymphocytes and leukocytes produce the major isoforms of APP, and after activation, they secrete amyloidogenic Aβ (Refs Reference Li15, Reference Allen16, Reference Mönning17, Reference Ledoux18).
Aβ1–42 has a higher ability to aggregate than the shorter Aβ1–40 (Refs Reference Jarrett and Lansbury19, Reference Kim and Hecht20). Aβ exists in soluble nontoxic monomers, strongly toxic soluble oligomers and in the form of less toxic insoluble fibrils. The soluble oligomers of Aβ1–42 are the most toxic (Refs Reference Lambert21, Reference Hartley22, Reference Walsh23, Reference Kirkitadze, Bitan and Teplow24, Reference Cerf25, Reference Pitt26). They form annular or pore-like structures that are indistinguishable from a class of pore-forming bacterial toxins (Refs Reference Kirkitadze, Bitan and Teplow24, Reference Cerf25, Reference Lashuel27), which cause rapid calcium influx through the targeted cell membranes (Refs Reference Rudel28, Reference Ayala29, Reference Müller30). Recent in vitro and in vivo studies showed that Aβ is an antimicrobial peptide (AMP) that targets bacterial membranes (Ref. Reference Soscia31). AMPs have proinflammatory activities and have a role in innate immune responses (Ref. Reference Soscia31).
Neurofibrillary tangles and neuropil threads contain paired helical filaments (PHFs) (Refs Reference Kidd32, Reference Terry33). The major component of PHFs is the microtubule-associated protein tau, which is in a pathological hyperphosphorylated state that abolishes microtubule assembly (Refs Reference Goedert, Trojanowski, Lee and Rosenberg34, Reference Lee35). Sequestration of peptidyl-prolyl cis/trans isomerase NIMA interacting 1 (PIN1) is one theory that explains the formation of these pathological fibrillar lesions (Ref. Reference Lu36).
Various hypotheses have been proposed to explain the pathogenesis of AD (Refs Reference Sherrington37, Reference Selkoe38, Reference Hardy39, Reference Tanzi and Bertram40, Reference Hardy and Selkoe41, Reference Bertram and Tanzi42, Reference Nagy43). A significant proportion of early-onset AD is inherited as an autosomal dominant trait (Ref. Reference Schellenberg44). Missense mutations of the APP gene located on chromosome 21 (Refs Reference Goate45, Reference Mullan46) are responsible for 5% of all early-onset familial AD cases (Ref. Reference Schellenberg44). Presenilin-1 (PS1) gene mutations are most frequent in early-onset familial AD (Refs Reference Sherrington37, 47). More than 80 different PS1 missense mutations or amino acid deletions have been identified (Refs Reference Sherrington37, 47, Reference Wasco48, Reference Perez-Tur49, Reference Miklossy50). Presenilin-2 (PS2) mutations are responsible for another subset of early-onset familial AD (Refs 47, Reference Levy-Lahad51). As originally shown by Hardy (Ref. Reference Hardy39), mutations in these pathogenic genes alter the processing of APP and result in an increase in amyloidogenic Aβ1–42 and Aβ1–43 (Refs Reference Miklossy50, Reference Citron52, Reference Cai, Golde and Younkin53, Reference Suzuki54, Reference Duff55, Reference Scheuner56, Reference Borchelt57, Reference Tomita58, Reference Citron59). The epsilon 4 allele of apolipoprotein E (APOE ɛ4) is an important risk factor for late-onset AD, which also correlates with increased Aβ burden (Ref. Reference Roses60). Finally, there is an association between AD and polymorphisms of various other genes, which include a growing number of genes, implicated in immune defence mechanisms (Refs Reference McGeer and McGeer61, Reference Bertram62).
The relation between the two major biological markers of AD, Aβ (Refs Reference Selkoe38, Reference Hardy and Selkoe41) and hyperphosphorylated tau (Refs Reference Goedert, Trojanowski, Lee and Rosenberg34, Reference Lee35), is not clear. Soluble Aβ and tau strongly interact (Ref. Reference Guo63), and APP is expressed in neurofibrillary tangles (Ref. Reference Perry64), suggesting that these apparently different pathologies are linked.
Alterations of various neurotransmitters, neuropeptides and hormones are reported to occur in AD (Refs Reference Davies, Katzman and Terry65, Reference Perry66, Reference Rossor67, Reference Fowler68). The cholinergic hypothesis is based on the alteration of acetylcholine synthesis, transport and release (Refs Reference Davies and Maloney69, Reference Bartus70). Oxidative damage to proteins, lipids and nucleic acids (Refs Reference Martins71, Reference Pappolla72, Reference Smith73, Reference Nunomura74) and mitochondrial dysfunction (Ref. Reference Parker75) are also significant contributors to the pathogenesis of AD. The role of various metals, including aluminium (Refs Reference Terry and Pena76, Reference Crapper, Krishnan and Dalton77) and iron (Ref. Reference Jellinger78) was proposed several decades ago. Direct modulation of APP processing by metal ions, including Ca2+, Zn2+, Fe2+/Fe3+ and Al3+, suggests that disrupted metal homeostasis also leads to increased APP levels (Ref. Reference Suh79). The calcium homeostasis hypothesis indicates that sustained deregulation of cytosolic calcium represents the common final pathway for neuronal death in AD (Ref. Reference Khachaturian80). Dysregulation of ubiquitylation or glycosylation processes (Refs Reference Mori, Kondo and Ihara81, Reference Lam82) has been shown in AD. Vascular lesions, including cerebral hypoperfusion and disturbed brain microcirculation, are also important factors (Refs Reference de la Torre and Mussivand83, Reference Esiri84, Reference Hachinski and Munoz85, Reference Snowdon86, Reference De Jong87, Reference Diaz88, Reference Kalaria89, Reference Suter90, Reference Miklossy91). Factors representing a risk for atherosclerosis (Refs Reference Muckle and Roy92, Reference Sparks93), are also risk factors for AD. Early involvement of the olfactory system in AD (Ref. Reference Averback94) led to the ‘olfactory hypothesis’, which suggests that putative pathogenic agents might access the brain by the olfactory pathways (Refs Reference Mann, Tucker and Yates95, Reference Hardy96, Reference Christen-Zaech97). Deregulation of various signalling pathways, apoptosis, craniocerebral trauma, exercise, environmental and nutritional factors, among others, are also implicated in the pathogenesis of AD.
The critical role of chronic inflammation and the importance of interleukin (IL)-1 signalling in AD is now widely recognised (Refs Reference McGeer98, Reference Griffin99, Reference McGeer and Rogers100). A series of inflammatory mediators, including cytokines, chemokines, proteases, adhesion molecules, free radicals, pentraxins, prostaglandins, anaphylatoxins and activated complement proteins, is present at the site of cortical lesions in AD (Refs Reference Schwab and McGeer101, Reference McGeer and McGeer102, Reference McGeer and McGeer103). The membrane attack complex (MAC, C5b-9) is also associated with plaques, tangles and neuropil threads (Refs Reference McGeer and Rogers100, Reference Webster104). Use of nonsteroidal anti-inflammatory drugs reduces the risk of AD (Refs Reference Stewart105, Reference Zandi106, Reference Veld107, Reference Akiyama108).
Nearly a century ago, Fischer, Alzheimer and their colleagues (Refs Reference Alzheimer2, Reference Fischer109) discussed the possibility that microorganisms could have a role in the formation of senile plaques. That a slow-acting unconventional infectious agent, acquired at an early age and requiring decades to become active, might be involved in AD was considered by several authors (Refs Reference Wisniewsky, Katzman, Terry and Bick110, Reference Khachaturian111). A growing number of recent observations indicate that infectious agents are involved in the pathogenesis of AD. Here, I review historical and recent observations on infectious agents related to AD and analyse the significance of the association and causal relationship.
Analogies between AD and the atrophic form of general paresis
Historical observations show that the clinical and pathological hallmarks defining AD are similar to those occurring in the atrophic form of general paresis, a chronic bacterial infection (Refs Reference Hübner112, Reference Perusini113, Reference Pacheco e Silva114, Reference Pacheco e Silva115, Reference Schlossberger, Brandis, Lubarsch, Henke and Roessle116, Reference Rizzo117, Reference Miklossy, Duyckaerts and Litvan118). In 1913, Noguchi and Moore (Ref. Reference Noguchi and Moore119) provided conclusive evidence that spirochetes are responsible for slowly progressive dementia, cortical atrophy and local amyloidosis.
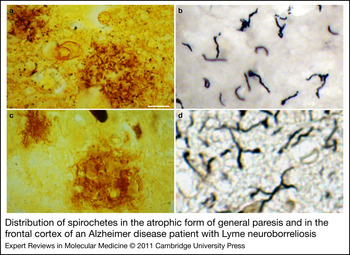
General paresis of the insane, paretic dementia or dementia paralytica is a chronic meningoencephalitis caused by the direct invasion of brain parenchyma by Treponema pallidum. Two forms are distinguished: the infiltrative and the atrophic form (Refs Reference Pacheco e Silva114, Reference Rizzo117). In the infiltrative form, mood disorders and psychosis predominate, and lymphoplasmocytic meningoencephalitis is the characteristic pathology (Refs Reference Rizzo117, Reference Miklossy, Duyckaerts and Litvan118). The atrophic form is characterised by slowly progressive dementia and cortical atrophy, which is accentuated in the frontotemporal regions (Refs Reference Pacheco e Silva114, Reference Pacheco e Silva115). Spirochetes form masses, plaques or colonies (Fig. 1) and disseminate as individual filaments, which are restricted to the cerebral cortex (Fig. 1) (Refs Reference Pacheco e Silva114, Reference Pacheco e Silva115). These spirochetal masses and individual spirochetes are morphologically identical to senile plaques (Fig. 1) and neuropil threads (Fig. 1). Pacheco e Silva (Refs Reference Pacheco e Silva114, Reference Pacheco e Silva115), reported that the number of spirochetes and spirochetal ‘plaques’, which are numerous in the hippocampus and frontal cortex, increases in parallel with the severity of cortical atrophy (Refs Reference Pacheco e Silva114, Reference Pacheco e Silva115). Lymphoplasmocytic infiltrates are absent. Severe neuron loss is accompanied by reactive microgliosis and astrocytosis and by accumulation of ‘paralytic iron’ (Ref. Reference Merritt, Adams and Solomon120). The occurrence of neurofibrillary tangles is also documented in general paresis (Refs Reference Perusini113, Reference Miklossy, Duyckaerts and Litvan118, Reference Bonfiglio121, Reference Vinken and Bruyn122), and the local amyloid (Ref. Reference Volland123), as in AD, consists of Aβ (Ref. Reference Miklossy, Rosemberg and McGeer124).

Figure 1. Distribution of spirochetes in the atrophic form of general paresis and in the frontal cortex of an Alzheimer disease (AD) patient with Lyme neuroborreliosis. Spirochetes form plaque-like masses (a,c) and disseminate as individual filaments (b,d) in general paresis (a,b) and AD (c,d), which are identical to senile plaques and curly fibres. (a,c) Bosma Steiner silver technique for the visualisation of spirochetes. (b) T. pallidum-specific polyclonal antibody. (d) Gallyas silver technique. Scale bars: 50 µm (a), 40 µm (c), 10 µm (b,d). All images were obtained from previously published studies: a (Ref. Reference Miklossy, Rosemberg and McGeer124); b (Ref. Reference Miklossy175); c, d (Ref. Reference Miklossy149).
Analogies between AD and other age-related chronic inflammatory disorders
Pathogens can produce slowly progressive chronic diseases. Following the pioneering work of Warren and Marshall (Ref. Reference Marshall and Warren125), it is today established that Helicobacter pylori causes stomach ulcers. Infectious agents are also linked to atherosclerosis, cardio- and cerebrovascular disorders (Refs Reference Laitinen126, Reference Saikku127, Reference Mendall128, Reference Martin de Argila129, Reference Renvert130, Reference Zaremba131, Reference Chiu132, Reference Haraszthy133, Reference Rassu134), chronic lung diseases (Refs Reference Martin135, Reference MacDowell and Bacharier136, Reference Micillo137), inflammatory bowel diseases and neuropsychiatric disorders (Refs Reference Marttila138, Reference Rott139, Reference Beaman and Caino140, Reference Gosztonyi and Ludwig141, Reference Salvatore142, Reference Langford and Masliah143).
Chlamydophila (Chlamydia) pneumoniae (Refs Reference Laitinen126, Reference Saikku127), H. pylori (Refs Reference Mendall128, Reference Martin de Argila129) and several periodontal pathogens, including invasive oral spirochetes (Refs Reference Renvert130, Reference Zaremba131) and herpes viruses, have been found in human atherosclerotic lesions. Some of them also enhanced atherosclerosis in experimental animals (Refs Reference Zaremba131, Reference Chiu132). These pathogens were also reported to be associated with AD (Refs Reference Balin144, Reference Balin145, Reference Miklossy146, Reference Riviere, Riviere and Smith147, Reference Miklossy148, Reference Miklossy149, Reference Jamieson150, Reference Itzhaki and Wozniak151).
Epidemiological studies have confirmed that several of these chronic inflammatory disorders are associated with AD (Refs Reference Olichney152, Reference Luchsinger153, Reference Voisin154, Reference Ott155). In addition, they are all linked to periodontal polybacterial disorders, which are primarily caused by Gram-negative bacteria (Refs Reference Kamer156, Reference Pihlstrom, Michalowicz and Johnson157, Reference Gibson158, Reference Mattila, Pussinen and Paju159). Spirochetes and herpes viruses are predominant periodontal pathogens (Refs Reference Seppaa and Ainamo160, Reference Ling161, Reference Heydenrijk162, Reference O'Brien-Simpson163), and C. pneumoniae is a major upper respiratory tract pathogen. An infectious origin might give a comprehensive explanation of the common cellular and molecular mechanisms, inflammatory processes and common inflammatory gene polymorphisms involved in these chronic inflammatory disorders and AD (Refs Reference McGeer and McGeer61, Reference DeGraba164, Reference Kolb and Mandrup-Poulsen165).
Evidence for the association of pathogens with AD
Spirochetes
Spirochetes are Gram-negative, helical bacteria, which possess endoflagella, taxonomically distinguishing them from other bacteria. There are over 200 different spirochetal species or phylotypes (Ref. Reference Dewhirst166). Spirochetes are causative agents of important human diseases such as syphilis, pinta, yaws, bejel, Lyme disease, Vincent angina, relapsing fever, leptospirosis, ulcerative gingivitis and various periodontal disorders (Ref. Reference Gastinel167). The major Borrelia species causing Lyme disease are B. burgdorferi (Ref. Reference Burgdorfer170), B. afzelii, B. garinii and B. valaisiana. Relapsing fever is caused by B. recurrentis. Oral spirochetes are predominant periodontal pathogens that are highly prevalent in the population and comprise diverse Treponema species (Refs Reference Dewhirst166, Reference Chan171). Several are invasive (Refs Reference Riviere172, Reference Peters173): they include T. denticola, T. socranskii, T. pectinovorum, T. amylovorum, T. lecithinolyticum, T. maltophilum, T. medium and T. putidum (Refs Reference Dewhirst166, Reference Chan171, Reference Wyss174). T. vincentii causes Vincent angina, a necrotising fusospirochetal disease (Ref. Reference Gastinel167).
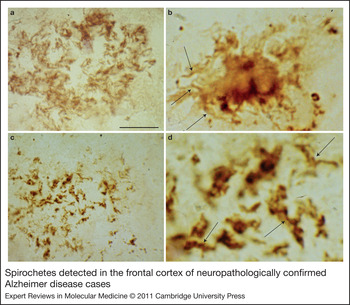
Because spirochetes are strongly neurotropic (Ref. Reference Gastinel167), it was expected that several types of spirochetes, in an analogous way to T. pallidum, might cause dementia, plaque- and tangle-like lesions, Aβ deposition and consequently might be involved in the pathogenesis of AD. To detect all types of spirochete, neutral techniques need to be used (Ref. Reference Miklossy146). Using dark-field microscopy, spirochetes were detected in the cerebrospinal fluid (CSF), in the blood and in the brain in 14 definite AD cases tested (Table 1). Spirochetes were not found in 13 age-matched controls without any AD-type cortical changes (Ref. Reference Miklossy146). Silver-stained helically shaped spirochetes were also detected by electron microscopy. Spirochetes were isolated from the cerebral cortex in these 14 AD cases (Table 1), and in three of them, they were cultivated from the brain in a selective medium for B. burgdorferi (Ref. Reference Miklossy146). Spirochetes were detected and isolated from the brains of eight additional AD cases derived from another laboratory and in the blood of five living patients with clinically diagnosed AD-type dementia (Ref. Reference Miklossy, Giacobini and Becker186; Table 1). Four healthy controls did not show spirochetes. Taxonomical analyses showed that the helically shaped microorganisms belong to the order Spirochaetales (Ref. Reference Miklossy187). To ensure the consistency of these results, the detection of spirochetes was also performed using various other techniques, including histochemistry, dark-field microscopy, atomic force microscopy, electron microscopy and immunoelectron microscopy, immunohistochemistry using spirochete and bacterial peptidoglycan (PGN)-specific antibodies (Refs Reference Miklossy146, Reference Riviere, Riviere and Smith147, Reference Miklossy149, Reference Miklossy, Giacobini and Becker186, Reference Miklossy187, Reference Miklossy176, Reference Miklossy175, Reference Miklossy188, Reference Miklossy189), and detection of specific and nonspecific bacterial DNA (Refs Reference Miklossy149, Reference Miklossy176). PGN is the building block of the cell wall of Gram-negative and Gram-positive bacteria; however, mycoplasma and chlamydiae lack detectable PGN (Refs Reference Hesse190, Reference McCoy191). The morphology of spirochetes detected by spirochete- or PGN-specific antibodies is identical (Fig. 2; compare also Fig. 7 G and H of Ref. Reference Miklossy175). PGN-immunoreactive spirochetes were detected in 32 definite AD cases and in 12 cases with mild or moderate AD-type cortical changes.

Figure 2. Spirochetes detected in the frontal cortex of neuropathologically confirmed Alzheimer disease cases. Spirochetes in an immature senile plaque detected by a cocktail of antibodies against Borrelia burgdorferi (a), in a mature plaque detected by in situ hybridisation (b, arrows) using Borrelia-specific probes, and in an amorphous plaque detected by antibacterial peptidoglycan antibody (c). (d) The central part of panel c at higher magnification. Arrows indicate helical spirochetes with the same morphology as observed in b. Scale bars: 80 µm (a), 30 µm (b), 80 µm (c), 20 µm (d). Images are from previously published studies: a,b (Ref. Reference Miklossy149); c,d (Ref. Reference Miklossy188).
Table 1. Detection of spirochetes in AD

Data reviewed in the literature with respect to the detection of all types of spirochetes using neutral techniques and the specific detection of periodontal pathogen Treponemas and Bb. Results of the statistical analysis are given for each group and for all studies together. AD indicates number of AD cases positive for spirochetes/ number of AD cases analysed; Control indicates number of control cases positive for spirochetes/number of control cases analysed. AD, Alzheimer disease; Bl, blood; CSF, cerebrospinal fluid; EM, electron microscopy; AFM, atomic force microscopy; DF, dark-field microscopy; HC, histochemistry (Warthin and Starry, Bosma-Steiner silver stain for spirochetes); IHC, immunohistochemistry; ISH, in situ hybridisation; PCR, polymerase chain reaction; Bb, Borrelia burgdorferi; Wbl, Western blot; P is exact value of the significance calculated by Fischer test; OR, odds ratio; CI, 95% confidence interval; +, positive; –, negative; ND, not determined; seq, sequencing.
aControls without any AD-type changes.
bCases from previous studies, which were subtracted when the total number of cases of studies was considered.
cCases with mild or moderate AD-type cortical changes.
dWhere the number of positive controls was zero, in order to calculate the OR and 95% CI, one positive case was added to the control group.
Other authors found no evidence of spirochetes in the brains of seven AD cases by dark-field or electron microscopy (Ref. Reference McLaughlin192). However, spirochetes were observed in the blood of one of 22 living patients with AD-type dementia (Table 1). The spirochete observed by these authors corresponded to the vegetative, regularly spiral form. They suggested that it could correspond to an oral spirochete. Whether the atypical, pleomorphic spirochetal forms, which commonly occur in infected tissues, blood and CSF (Refs Reference Gastinel167, Reference Miklossy175, Reference Jacquet and Sézary177, Reference Mattman193), were considered by the authors is not clear.
Periodontal pathogen Treponemas
Using molecular and immunological techniques, six of seven periodontal Treponema species, namely T. socranskii, T. pectinovorum, T. denticola, T. medium, T. amylovorum and T. maltophilum, were identified in the brains of AD patients using species-specific polymerase chain reaction (PCR; Table 1). At least one oral Treponema species was present in 14 of 16 AD brains, compared with 4 out of 18 controls (Ref. Reference Riviere, Riviere and Smith147). T. pectinovorum and T. socranskii antigens were observed in 15 of 16 AD brains and in 7 of 18 controls. In the hippocampus and in the frontal cortex. Six different Treponema species were detected in one AD patient, five species in four, four or three species each in one AD case, and one species in seven AD brains. Two Treponema species were observed in one control and one species in the other three positive controls. These results reinforce previous observations (Ref. Reference Miklossy146) and indicate that these periodontal pathogen spirochetes, in an identical way to T. pallidum and B. burgdorferi, have the ability to invade the brain, persist in the brain, and cause dementia, cortical atrophy and amyloid deposition.
Borrelia burgdorferi
The causative agent of Lyme disease is transmitted by the bite of infected ticks (Ref. Reference Burgdorfer170). Neurological complications occur in about 15% of affected individuals. Dementia and subacute presenile dementia both occur in Lyme disease (Refs Reference Miklossy146, Reference Miklossy149, Reference MacDonald194, Reference Dupuis195, Reference Schaeffer196, Reference Fallon and Nields197, Reference Pennekamp and Jaques198). B. burgdorferi was first detected in the brains of two AD patients (Table 1) by MacDonald and Miranda (Ref. Reference MacDonald and Miranda178) and MacDonald (Ref. Reference MacDonald199). This spirochete was detected in the cerebral cortex by dark-field microscopy and with a specific antibody against B. burgdorferi. This species was also detected and cultivated from the brains of three definite AD cases in an initial series of 14 AD cases (Ref. Reference Miklossy146). Molecular characterisation, using 16S rRNA gene sequence analysis, identified these spirochetes as B. burgdorferi sensu stricto (s.s.) (Ref. Reference Miklossy149). Electron microscopy analysis confirmed that these spirochetes possess 10–15 endoflagella typical of Borrelia species. Two of the three AD patients had a positive CSF serology for B. burgdorferi, and the 31 kDa outer surface protein A (OspA) band, which is highly specific for B. burgdorferi, was detected by western blot in these three AD cases (Ref. Reference Miklossy149). The pathological changes found in the brain were identical to those occurring in the atrophic form of general paresis (Ref. Reference Miklossy149). The cortical distribution of spirochetes in masses or colonies was identical to those of senile plaques, and the morphology of individual spirochetes was identical to those of curly fibres (Ref. Reference Miklossy149). B. burgdorferi antigens colocalised with Aβ in cortical plaques and in leptomeningeal and cortical arteries containing amyloid deposits. Neurofibrillary tangles were also immunoreactive for B. burgdorferi. OspA and flagellin genes were detected in AD-type lesions using in situ hybridisation (ISH) (Fig. 2b). In additional AD patients with concurrent Lyme neuroborreliosis, B. burgdorferi-specific antigens (Ref. Reference Miklossy146) and DNA (Ref. Reference Meer-Scherrer179) were observed. Using species-specific PCR, B. burgdorferi DNA was detected in 5 of 16 AD patients tested and in 1 of 18 controls, all of which also had oral Treponema spirochetes (Ref. Reference Riviere, Riviere and Smith147). Finally, B. burgdorferi-specific DNA was detected by both ISH and PCR in the hippocampus in 7 of 10 pathologically confirmed AD cases (Refs Reference MacDonald183, Reference MacDonald184) (Table 1).
Pappolla and colleagues (Ref. Reference Pappolla185) failed to detect B. burgdorferi in six AD patients. They stated that they could not exclude other spirochetes not detected by their methods. In all other studies where B. burgdorferi was not detected, evidence is lacking on whether the analysed AD patients had Lyme neuroborreliosis or not (Refs Reference Gutacker180, Reference Marques181) (Table 1). Similarly, the analysis of the serology of B. burgdorferi alone, as a result of the low incidence of Lyme dementia compared with AD, might give false-negative results (Refs Reference Gutacker180, Reference Galbussera182). To demonstrate the role of B. burgdorferi, AD patients with Lyme neuroborreliosis should be analysed.
Taken together, these observations show that various authors detected and cultivated various types of spirochetes from the brains of AD patients. Coinfection with several types of spirochetes occurs.
Chlamydophyla pneumoniae
Several authors reported the existence of an association between C. pneumoniae, an obligate intracellular respiratory pathogen, and AD (Refs Reference Balin144, Reference Gérard200, Reference Gérard201, Reference Paradowski202, Reference Appelt203) (Table 2). C. pneumoniae-specific DNA was detected in the brains in 90% of sporadic AD patients and in 5% of controls (Ref. Reference Balin144) (Table 2). Two mRNAs, encoding KDO transferase and a 376 kDa protein, specific to C. pneumoniae, were also identified in frozen AD brain tissue by reverse transcriptase-PCR (RT-PCR).
Table 2. Detection of Chlamydophyla pneumoniae and other bacteria in AD

AD, number of AD cases with positive detection/number of AD cases analysed; Control, number of control cases with positive detection/number of control cases analysed; CSF, cerebrospinal fluid; PCR, polymerase chain reaction; RT-PCR, reverse transcriptase-PCR; EM, electron microscopy; IHC, immunohistochemistry; Cult, culture. P, exact value of significance following Fischer test; OR, odds ratio; CI, 95% confidence interval values. aPositive in at least one of several samples.
Immunohistochemical analyses of AD brains showed C. pneumoniae in microglia, perivascular macrophages and astrocytes, and in about 20% of neurons (Refs Reference Balin144, Reference Gérard200, Reference Gérard201, Reference Appelt203, Reference Hudson and Dolmer207, Reference MacIntyre211). They were commonly found in brain regions showing the characteristic neuropathology of AD (Refs Reference Balin144, Reference Gérard200, Reference Gérard201). The presence of C. pneumoniae in the brains of AD patients was also confirmed by immunoelectron microscopy using specific monoclonal antibody against the outer membrane protein of C. pneumoniae. Electron and immunoelectron microscopy identified both chlamydial elementary bodies and reticulate bodies (RBs) (Refs Reference Albert212, Reference Balin144). That the replicative RB form was also detected in glial cells, neurons and pericytes indicates that a viable and transcriptionally active form of the microorganism is present in these cells (Refs Reference Gérard201, Reference Arking213, Reference Dreses-Werringloer214, Reference Dreses-Werringloer215). Pleomorphic forms of C. pneumoniae were also observed (Ref. Reference Arking213). C. pneumoniae-specific DNA was detected in the CSF in a significantly higher number of cases in AD patients (43.9%) than in controls (10.6%) (Ref. Reference Paradowski202). C. pneumoniae was cultured from various brain samples of AD patients originating from different geographic regions of North America (Refs Reference Balin144, Reference Gérard201, Reference Dreses-Werringloer215) and was also isolated from the CSF (Ref. Reference Paradowski202). Tor-1 and Phi-1 isolates were characterised by PCR assays targeting C. pneumoniae-specific genes Cpn0695, Cpn1046 and tyrP. Two groups, using paraffin-embedded brain samples, failed to detect C. pneumoniae in AD or in controls by PCR (Refs Reference Nochlin216, Reference Gieffers217). In another study, C. pneumoniae was detected in 2 of 15 AD cases and in 1 of 5 controls (Ref. Reference Ring and Lyons204).
Other bacteria
Propionibacterium acnes, an atypical anaerobic bacterium, was identified in biopsy specimens of the frontal cortex in three of four AD patients and in one of five controls with cerebral tumour. The P. acnes positive control was an elderly patient with cardiovascular risk factors and glioblastoma (Ref. Reference Kornhuber205). P. acnes was identified by microbiological methods and by gas chromatography. The bacterium was cultivated from frontal cortical biopsy specimens in Schaedler blood agar, at 35°C, under anaerobic conditions (Refs Reference Kornhuber205, Reference Kornhuber206). P. acnes has long been considered to be a commensal bacillus of the skin. Recent observations showed that P. acnes, the causative agent of acne vulgaris, is implicated in various infections, including brain abscesses, endocarditis, endophthalmitis and osteomyelitis (Refs Reference Delahaye208, Reference Brook209). P. acnes was also shown to be a predominant periodontal pathogen (Ref. Reference Preza218). By haematogenous dissemination, it can reach and infect various organs, including the brain. Stabilisation of the clinical symptoms and memory improvement were observed in two AD cases treated with P. acnes-sensitive cephalosporine combined with enalapril and oestrogen (Ref. Reference Kornhuber206). The author pointed to microangiopathy as the underlying pathology (Refs Reference Kornhuber219, Reference Kornhuber220). Because the number of AD cases analysed is low, further studies should be encouraged to determine any association of P. acnes with AD.
Actinomycetes have also been suggested to be involved in AD, with an incidence four times higher than in other pathological conditions (Ref. Reference Howard and Pilkington221). Ultrastructural analysis revealed that the fibronectin-immunopositive fibrillary lesions in senile plaques, which were negative for neuronal, glial and macrophage markers, are compatible with filamentous microorganisms and might correspond to Actinomycetes (Ref. Reference Howard and Pilkington221). It is noteworthy that Actinobacillus actinomycetemcomitans is a frequent periodontal pathogen (Ref. Reference Fenesy222) and that Nocardia asteroides was reported to cause Parkinson-like symptoms in experimental animals (Ref. Reference Kohbata and Beaman223).
Finally, the causative agent of stomach ulcers, H. pylori (Ref. Reference Marshall and Warren125), has also been suggested to be associated with AD (Refs Reference Malaguarnera224, Reference Kountouras225, Reference Kountouras226). Serum IgG and IgA antibodies against H. pylori occurred in a higher percentage in the group of 30 AD patients compared with 30 controls (Ref. Reference Malaguarnera224). The difference is statistically significant as determined by post-hoc comparison (Ref. Reference Honjo, van Reekum and Verhoeff210). An almost twofold higher prevalence of gastric H. pylori infection was observed in clinically diagnosed AD patients and in patients with mild cognitive decline (Refs Reference Kountouras225, Reference Kountouras227) than in controls. H. pylori-specific IgG antibody levels were also significantly higher in the blood and CSF of AD patients than in controls (Ref. Reference Kountouras226). Additional studies would be of interest in detecting whether H. pylori is present in the brain and to analyse the possibility of a causal relationship between H. pylori and AD.
Herpes simplex virus type-1 (HSV-1) and other viruses
HSV-1 is a common neurotropic virus that infects around 70% of the population after the age of 50 (Refs Reference Jamieson150, Reference Jamieson228, Reference Bertrand229, Reference Xu230). HSV-1 DNA was detected (Table 2) in brain samples using ISH in some elderly patients with dementia (Ref. Reference Sequiera231). Three other studies failed to detect HSV-1 by ISH in AD or in control brains (Refs Reference Middleton232, Reference Pogo and Elizan233, Reference Pogo, Casals and Elizan234, Reference Walker, O'Kusky and McGeer235). Increasing numbers of recent observations have detected HSV-1 DNA in the brain in AD (Refs Reference Jamieson150, Reference Itzhaki and Wozniak151, Reference Jamieson228, Reference Bertrand229, Reference Itzhaki236, Reference Itabashi237, Reference Lin238). Using PCR, Jamieson et al. (Ref. Reference Jamieson150) observed HSV-1 DNA in the brains of a high proportion of elderly subjects, with or without AD, which was absent or less frequent in young controls (Ref. Reference Jamieson228). Several authors showed that HSV-1 is a significant risk factor (Table 3) when present in AD patients who are carriers of APOE ɛ4 (Refs Reference Itzhaki236, Reference Itabashi237, Reference Lin238), but this is not supported by Beffert et al. (Refs Reference Beffert239, Reference Beffert240). In situ PCR showed that HSV-1 DNA is localised to senile plaques (Ref. Reference Renvoize, Awad and Hambling243). Ninety per cent of senile plaques in AD and 80% in normal ageing subjects contained viral DNA (Ref. Reference Wozniak, Mee and Itzhaki241).
Table 3. Detection of HSV-1 in AD

Number of Alzheimer cases with positive detection/number of Alzheimer cases analysed; Control, number of control cases with positive detection/number of control cases analysed. P, value of significance following Fischer test; OR, odds ratio; CI, 95% confidence interval values; +, positive; –, negative; AD, Alzheimer disease; CSF, cerebrospinal fluid; HSV-1, herpes simplex virus type-1; IHC, immunohistochemistry; ISH, in situ hybridisation; PCR, polymerase chain reaction; APOE ɛ4: epsilon 4 allele of apolipoprotein E.
Anti-HSV-1 antibodies as detected in the CSF by enzyme-linked immunosorbent assay (ELISA) were also significantly higher in AD patients than in younger controls, but without a significant difference between the AD and age-matched control groups (Ref. Reference Wozniak242). Increased titres of HSV-1 IgGs, which characterise past infection, were observed in AD cases and age-matched controls, without a significant difference between the two groups (Refs Reference Renvoize, Awad and Hambling243, Reference Ounanian244). In a large prospective study, in addition to HSV-1 IgG, the presence of IgM, which characterises active primary infection or reactivation of the infection, was also assessed in the sera of 512 elderly patients, initially free of dementia. During 14 years of follow-up, 77 AD cases were diagnosed. In contrast to IgG, IgM-positive subjects showed a significantly higher risk of developing AD, indicating that reactivation of HSV-1 seropositivity is correlated with AD (Ref. Reference Letenneur245).
Other herpes viruses were also detected in the brain using PCR: human herpes virus 6 (HHV6) types A and B, herpes simplex virus type-2 (HSV-2) and cytomegalovirus (CMV). HHV6, HSV-2 and CMV were observed in 70%, 13% and 36% of AD patients and in 40%, 20% and 35% of controls, respectively. The differences between the groups were not statistically significant (Ref. Reference Lin246).
In addition to CMV, a possible association between HLA-BW15 and AD has also been reported (Ref. Reference Renvoize247). A recent study revealed that elderly subjects with high levels of antibody against CMV develop more severe cognitive decline over 4 years (Ref. Reference Aiello248) compared with controls.
The adenovirus early region 1A (E1A) gene and its expression using ISH and immunohistochemistry were analysed in five AD cases and in two controls. Reactive microglial cells in both AD (5/5) and control brain tissue (2/2) showed positive hybridisation and immunoreactive expression of adenovirus E1A, indicating a monocyte- or microglia-mediated entry of adenovirus into the central nervous system (CNS) (Ref. Reference Matsuse249).
Borna disease virus (BDV) was linked to affective disorders and schizophrenia (Refs Reference Rott139, Reference Salvatore142). A few attempts have been made to analyse the prevalence of BDV antibodies and BDV p40 gene coding sequences in AD (Refs Reference Igata250, Reference Yamaguchi251, Reference Chalmers, Thomas and Salmon252), which did not show an association (Table 3). Following Borna-virus-induced infection in APP(Tg2576) transgenic mice, a reduction of cortical and hippocampal Aβ deposits was observed (Ref. Reference Stahl253). One explanation is that the stimulation and activation of microglia might produce this effect.
The human immunodeficiency virus type-1 (HIV-1) (Ref. Reference Rempel and Pulliam254) is able to induce the biological hallmarks of AD; however, the virus is present in the brains of mostly young patients suffering from acquired immune deficiency syndrome (AIDS). The virus invades mostly macrophages and glial cells and the virus itself does not reproduce the pathological hallmarks of AD. However, by affecting immune defences, HIV-1 facilitates infection by various pathogens, including, among others, HSV-1, CMV, C. pneumoniae and spirochetes (Refs Reference Lang255, Reference Hook256, Reference Comandini257, Reference Tobian and Quinn258).
Correlates of infection risk and AD
Based on the data available on the association of spirochetes, C. pneumoniae and HSV-1 with AD, contingency tables were used to analyse the strength of the association and the risk of infection in AD. In those studies where all types of spirochetes were detected using neutral techniques, spirochetes were observed in the brain in 90.1% (64/71) of AD cases and were absent in controls without any AD-type changes. This difference is significant (Table 1), and the association remains significant when cases where spirochetes were analysed in the blood are also included. The association between periodontal pathogen spirochetes and AD is also statistically significant. They were detected in the brain in 93.7% of AD cases and in 33.3% of controls. B. burgdorferi was about 13 times more frequent in AD cases than in controls, a statistically significant difference. It is noteworthy that B. burgdorferi was detected in all AD cases with a positive serology or where spirochetes were cultivated from the brain (Table 1). Taken together, in all studies where spirochetes or their specific species (periodontal Treponema spirochetes or B. burgdorferi) were detected in the brain, it can be concluded that the frequency of spirochetes is more than eight times higher in AD cases (90/131; 68.7%) than in controls (6/71; 8.41%). That spirochetes were cultivated from the brains of AD patients indicates that viable spirochetes are present in advanced stages of dementia. They can sustain persisting infection and inflammation and cause neuronal destruction (Ref. Reference Miklossy148).
The frequency of C. pneumoniae was about five times higher in AD cases (60/125; 48%) than in controls (5/52; 9.6%). The difference remains significant when those cases where C. pneumoniae was detected in the CSF were also included (Table 1). That C. pneumoniae was cultivated from the brain (Refs Reference Gérard200, Reference Arking213) and CSF (Ref. Reference Paradowski202) and that replicative RBs were present in glial cells, neurons and pericytes in AD indicate that this microorganism is also present in a viable, active form in the brain in AD.
There is no significant difference between the frequency of HSV-1 detected in the brain in AD patients compared with age-matched control. However, a significant difference was observed between APOE ɛ4-positive and -negative AD carriers, as reported by several authors (Refs Reference Itzhaki236, Reference Itabashi237, Reference Lin238, Reference Burgos259, Reference Miller and Federoff260), except for Beffert and colleagues (Refs Reference Beffert239, Reference Beffert240). A significant association of AD with ongoing or reactivated HSV-1 infection was reported (Ref. Reference Letenneur245).
Evidence for a causal role of pathogens in AD
Additional studies have brought further evidence in favour of a probable causal relationship between spirochetes, C. pneumoniae, HSV-1 and AD. Exposure of primary mammalian neuronal and glial cells to spirochetes, namely to B. burgdorferi, which can be cultivated and propagated in pure culture, generated thioflavin-S-positive and Aβ-immunoreactive amyloid plaques and tangle- and granulovacuolar-like formations in vitro (Refs Reference Miklossy175, Reference Miklossy261). In situ, in the spirochete-induced Aβ plaques, synchrotron infrared microspectroscopy analysis revealed the presence of a β-pleated sheet conformation (Ref. Reference Miklossy261). Spirochete-induced increases in Aβ, APP and phosphorylated tau were all detected by western blot in infected cell cultures (Ref. Reference Miklossy261). This additional experimental evidence indicates that spirochetes are able to induce an AD-type host reaction and reproduce the defining pathological and biological hallmarks of AD (Refs Reference Miklossy175, Reference Miklossy261). Similar in vitro studies performed on CNS organotypic cultures, which aim to replace in vivo experiments, showed identical results (Ref. Reference Miklossy261). Reference B. burgdorferi spirochetes (strain B31) and those cultivated from the AD brain (strains ADB1 and ADB2) invaded neurons and glial cells and induced nuclear fragmentation in vitro, indicating that the spirochetes cultivated from the brains of AD patients are invasive and cause neuronal and glial damage and apoptosis (Refs Reference Miklossy175, Reference Miklossy261). Spirochetes occur in both extra- and intracellular locations. They invade neurons in vitro, in primary neuronal cultures (Refs Reference Miklossy175, Reference Miklossy261), in the cerebral cortex and in the trigeminal ganglia of AD patients (Refs Reference Miklossy146, Reference Riviere, Riviere and Smith147, Reference Miklossy148, Reference Miklossy149, Reference Miklossy175, Reference MacDonald183, Reference MacDonald184, Reference Miklossy261). Historical observations and illustrations showing that chronic spirochetal infection can reproduce the clinical, pathological and biological hallmarks of AD strongly support a causal relationship between spirochetal infection and AD (Refs Reference Pacheco e Silva114, Reference Pacheco e Silva115, Reference Miklossy, Rosemberg and McGeer124). All these observations indicate that various types of spirochetes, including B. burgdorferi and several periodontal pathogen spirochetes, in an analogous way to T. pallidum, can cause dementia, cortical atrophy and the pathological and biological hallmarks of AD.
Astrocytes and microglia infected in vitro with C. pneumoniae display inclusions that are indistinguishable from those characteristic of active infection of the standard HEp-2 host cell line (Refs Reference Dreses-Werringloer214, Reference Dreses-Werringloer215). It was reported that chronic or persistent infection of CNS cells with C. pneumoniae can affect apoptosis in AD, in both a pro- and antiapoptotic manner (Refs Reference Appelt203, Reference Dreses-Werringloer214, Reference Dreses-Werringloer215). Furthermore, infection of BALB/c mice by intranasal inhalation of C. pneumoniae initiated Aβ1–42 deposits in the brain that resembled senile plaques (Ref. Reference Little262). Antibiotic treatment following C. pneumoniae infection limited the number of induced amyloid plaques in vivo (Ref. Reference Hammond, Igbal, Winblat and Avila263).
The glycoprotein B (gB) of HSV-1 has a highly homologous sequence to a fragment of Aβ (Ref. Reference Cribbs264). Synthetic peptides derived from this region accelerate fibrillar aggregation of Aβ in vitro. They can self-assemble into fibrils, which are ultrastructurally indistinguishable from Aβ and are neurotoxic at a similar dose to Aβ. It was proposed that HSV-1 might act as a ‘seed’ for senile plaque formation (Ref. Reference Satpute-Krishnan, DeGiorgis and Bearer265). It was also shown that HSV-1 is associated with APP during its anterograde transport, which might affect APP degradation and synaptic function (Ref. Reference Satpute-Krishnan, DeGiorgis and Bearer265).
Exposure of cultured cells to HSV-1 results in increased intracellular Aβ levels in neurons and glial cells as analysed by immunocytochemistry, ELISA and western blot. Tau phosphorylation was also observed at a number of sites that are phosphorylated in AD (Refs Reference Wozniak266, Reference Zambrano267, Reference Wozniak, Frost and Itzhaki268). It was suggested that the association of viral DNA and senile plaques is very likely to be causal, because HSV-1 is able to increase the level of Aβ in neurons of infected mice in vivo (Ref. Reference Wozniak266).
Evidence for underlying mechanisms
Sources and dissemination
Spirochetes, C. pneumoniae and HSV-1 are all able to invade the brain (Fig. 3) and generate latent and persistent chronic infection (Refs Reference Balin144, Reference Balin145, Reference Miklossy146, Reference Riviere, Riviere and Smith147, Reference Miklossy148, Reference Miklossy149, Reference Jamieson228, Reference Bertrand229, Reference Leinonen269). The strong neurotropism of spirochetes is well known (Ref. Reference Gastinel167). In addition to haematogenous dissemination, spirochetes can spread through the lymphatic system and along nerve fibre tracts (Ref. Reference Gastinel167). Periodontal invasive spirochetes were detected in the trigeminal ganglia and along the trigeminal nerve (Ref. Reference Riviere, Riviere and Smith147). They might also propagate along the fila olfactoria and tractus olfactorius, which would enable them to reach the CSF, the septal and hippocampal regions in the earliest stages of the disease. This would be in harmony with the olfactory hypothesis (Refs Reference Averback94, Reference Mann, Tucker and Yates95, Reference Hardy96) and the early involvement of the olfactory tract and bulb (Ref. Reference Christen-Zaech97).

Figure 3. Sources and dissemination of pathogens associated with Alzheimer disease.
Through infected circulating monocytes, C. pneumoniae can also spread by haematogenous dissemination and, by crossing the blood–brain barrier, infect the brain. C. pneumoniae is an upper respiratory tract pathogen and can reach the brain through the olfactory system (Ref. Reference Mann, Tucker and Yates95). Intranasal inhalation of C. pneumoniae initiated plaque-like Aβ1–42 deposits in the brain in BALB/c mice, and C. pneumoniae-specific DNA was detected by PCR in the olfactory bulb in AD (Ref. Reference Little262).
HSV-1 infection might also reach the brain through the olfactory system, by infection of cranial and peripheral nerves and their ganglia and through haematogenous dissemination (Refs Reference Boggian270, Reference Valyi-Nagy271). The virus can reside in the brain in a latent form and be reactivated by peripheral infection, stress or immunosuppression (Ref. Reference Wozniak, Frost and Itzhaki268).
Neuroinflammation and TLR signalling
Persisting, poorly degradable bacterial remnants in mammalian tissues act as chronic inflammatory stimuli (Refs Reference Fox272, Reference Stimpson273, Reference Fleming, Wallsmith and Rosenthal274). Lipopolysaccharides (LPSs), PGN and various bacterial lipoproteins elicit a variety of proinflammatory responses and might represent an important source of inflammation in AD. Complex interactions between the innate and adaptive immune systems have a major role in infection (Ref. Reference Palaniyar275). The functions of innate immunity allow host cells to recognise most microorganisms, execute proinflammatory defences and start adaptive immune responses.
Bacteria attach to host cells through a variety of cell-surface components, including surface amyloid proteins, which interact with host proteases (Refs Reference Gebbink276, Reference Hammer277, Reference Gophna278, Reference Ben Nasr279, Reference Olsén, Jonsson and Normark280, Reference Sjöbring, Pohl and Olsén281, Reference Hammar282, Reference Umemoto, Li and Namikawa283). Proteolysis of the extracellular matrix allows bacteria to penetrate the basement membrane and invade host cells.
It is the innate immune system that provides the first line of defence against microorganisms. Pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), recognise unique structures of invading microorganisms (Ref. Reference Crack and Bray284). Patients with genetic defects related to signalling pathways activated by TLRs frequently suffer from severe recurrent infection (Refs Reference Lorenz285, Reference Kiechl, Wiedermann and Willeit286, Reference Akira and Takeda287, Reference Tobias and Curtiss288, Reference Turvey and Hawn289). In the absence of TLRs, death from experimental sepsis is significantly enhanced (Ref. Reference Akira and Takeda287). Various bacteria, including spirochetes, activate TLR signalling and interact with CD14 (Refs Reference Bulut290, Reference Sellati291, Reference Schroeder292). T. pallidum and B. burgdorferi or their synthetic membrane lipoproteins are major inflammatory mediators (Refs Reference Radolf293, Reference Ramesh294) and activate both the classical and alternative complement pathways, which opsonise and kill pathogens without the need for antibody.
Complex interactions between pathogen-associated molecular patterns (PAMPs), PRRs and TLR signalling pathways have a major role in linking innate and adaptive immunity and maintaining pathogen-free host tissues (Ref. Reference Palaniyar275). TLRs activate two major signalling pathways (Fig. 4). The core pathway activated by most TLRs leads to the activation of transcription factor nuclear factor-kappa B (NF-κB) and the mitogen-activated protein kinases (MAPKs) p38 and Jun kinase (JNK). The second pathway is activated by TLR3 and TLR4 and results in the activation of both NF-κB and interferon regulatory factor-3 (IRF3), allowing the induction of another set of inflammatory genes, including the antiviral interferon-β gene (IFNB).

Figure 4. Schematic representation of signalling pathways implicated in host–pathogen interactions in Alzheimer disease. Pattern recognition receptors (PRRs) recognise conserved structural components of microorganisms, called pathogen-associated molecular patterns (PAMPs) or ligands, which include peptidoglycan (PGN), lipoteichoic acid (LTA), flagellin (FLA), bacterial lipoprotein (BLP) and nucleic acid structures, such as bacterial CpG DNA or viral RNA, unique to bacteria and viruses. Lipopolysaccharide (LPS) is recognised following its binding to lipoprotein binding protein (LBP). Only Toll-like receptors (TLRs) have a cytoplasmic domain for signal transduction. Plasma-membrane-localised TLRs include TLR1, TLR2, TLR4, TLR5, TLR6 and TLR10, whereas endosomal TLRs include TLR3, TLR7, TLR8 and TLR9. TLR9 is responsible for the recognition of CpG islands of bacterial DNA. Complex interactions between pathogen-associated molecular patterns (PAMPs), pattern recognition receptors (PRRs) and TLR signalling pathways have a major role in linking innate and adaptive immunity and maintaining pathogen-free host tissues. Mammals use antimicrobial peptides (AMPs) as part of their innate immune defences to destroy invading pathogens. In contrast, in a similar way, pore-forming bacterial surface proteins are also able to destroy host cells. Evasion of pathogens from destruction by the host immune defences will result in chronic persistent infection and host cell destruction. APP, amyloid precursor protein; Aβ, amyloid beta; CD14, cluster of differentiation 14; NF-κB, nuclear factor-kappa B; MALP2, mycoplasma diacylated lipopeptide 2; p38 MAPK, mitogen-activated protein kinase; JNK, c-Jun kinase; MAC, membrane attack complex; MD2, lymphocyte antigen 96; Akt, serine/threonine protein kinase Akt1; IRF, interferon regulatory factor; IL, interleukin; TNF, tumour necrosis factor; COX2, cyclooxygenase 2; IFN, interferon, CREB, cAMP response element binding.
Activation of p38 MAPKs during bacterial infection was also shown to be crucial in the local production of cytokines such as IL-8 (Ref. Reference Ratner295) and in the development of effective immune responses in vivo (Ref. Reference van den Blink296). Toxin-induced p38 MAPK activation requires pore formation and is inhibited by the relief of osmotic stress.
Pore-forming toxins are the most common class of bacterial protein toxins and are often important virulence factors (Ref. Reference Van der Goot297). Pore-forming bacterial toxins are typically oligomers of soluble, monomeric proteins or peptides, which form transmembrane channels. Channel formation in the membrane of targeted cells, which triggers cellular ion imbalance, is a widely used form of bacterial attack (Refs Reference Gekara298, Reference Gonzalez299). These pore-forming bacterial toxins generate calcium-dependent and lipid-mediated signalling on host cell surfaces, leading to a variety of events such as tyrosine phosphorylation (Ref. Reference Gekara and Weiss300), actin rearrangement (Ref. Reference Cossart and Lecuit301), NF-κB activation (Ref. Reference Kayal302) and regulation of gene expression through histone modification (Ref. Reference Hamon303).
Mammals also use bacterial porin-like proteins such as perforins as part of their innate immune defences (Ref. Reference Pipkin and Lieberman304). Aβ1–42 was shown to be an AMP that belongs to the innate immune system. Toxic oligomers of Aβ1–42 bind to lipid bilayers of bacterial membranes and enveloped viruses, triggering Ca2+ influx and bacteriolysis or viral destruction. Deregulation of the balance between host cell destruction by pathogens and pathogen destruction by the host immune systems will influence the outcome of infections.
The innate and alternative pathways through the common membrane attack complex (MAC, C5b9) cause bacteriolysis (Refs Reference Blanco305, Reference Lawrenz306, Reference Kraiczy307). Cellular and humoral components of the immune system reactions are both associated with AD (Refs Reference McGeer98, Reference Griffin99, Reference McGeer and Rogers100, Reference Schwab and McGeer101, Reference McGeer and McGeer102). The adaptive immune system kills pathogens through the formation of specific antibodies directed against invading microorganisms. Monocytes, macrophages and microglia participate in both the innate and adaptive immune responses. When activated, they secrete chemokines and cytokines and express various proinflammatory molecules needed for the efficient removal of pathogens and damaged cells.
Neuroinflammatory responses that include activation of glia with marked elevation of cytokine expression, in particular elevation of IL-1, are observed in AD, in Down syndrome fetuses and children before plaque formation, and also in HIV infection (Refs Reference Griffin99, Reference Stanley308, Reference Brabers and Nottet309). IL-1 directs both the in vivo (Ref. Reference Sheng310) and in vitro synthesis of APP (Ref. Reference Goldgaber311), thus favouring senile plaque formation in AD. IL-1 upregulation of ApoE promotes ApoE-mediated induction of APP (Ref. Reference Barger312) and MAPK-p38-mediated tau phosphorylation, in part by upregulation of MAPK-p38 synthesis and activation and by favouring the formation of neurofibrillary tangles (Refs Reference Li313, Reference Sheng314, Reference Sheng315). IL-1 also influences neurotransmission because it increases the synthesis and activation of AChE (Ref. Reference Li316) and induces neuron loss by increasing caspase 1 (IL-1β cleavage enzyme) activity (Refs Reference Zhu317, Reference Li318). Microglial activation by APP and release of secreted APP (sAPP) modulated by apolipoprotein E also have an important role in the degenerative process of AD (Refs Reference Li313, Reference Barger and Harmon319). Activation of microglia with TLR2, TLR4 and TLR9 ligands increases Aβ ingestion in vitro (Ref. Reference Tahara320). Furthermore, stimulation of the immune system through TLR9 in APP transgenic (Tg2576) mice reduces Aβ deposition (Ref. Reference Scholtzova321).
Evasion and establishment of latent and chronic infection
The ability of spirochetes and C. pneumoniae to evade destruction by host immune systems and establish chronic infection is well known. HSV-1 can also remain in a latent state and persist throughout life in human tissues. In the latent state, the viral genome is present, but no viral particles are produced. Viral gene expression during latency is limited to one locus – the gene encoding the latency-associated transcript (Ref. Reference Itzhaki and Wozniak151).
Pathogens use a broad range of strategies to overcome antigenic recognition, complement lysis and phagocytosis. Blockade of the complement cascade or acquisition of host-derived complement inhibitors results in their evasion from complement lysis. This allows microbial survival and proliferation even in immune-competent hosts. Complement-resistant strains of B. burgdorferi possess five complement regulatory acquiring surface proteins, which bind host complement inhibitors (Refs Reference Kraiczy307, Reference Kraiczy and Würzner322) and a CD59-like molecule (Refs Reference Kraiczy and Würzner322, Reference Pausa323). A fragment of HSV-1 glycoprotein C (gC) shares sequence similarity with host complement receptor 1 (CR1), showing that viruses can also escape from attack by the MAC. Bacteria and viruses protect themselves from destruction by the host adaptive immune system. B. burgdorferi induces IL-12, a cytokine recently recognised to be critical for driving cellular responses towards the Th1 subset of T helper cells (Refs Reference Radolf293, Reference Rasley, Anguita and Marriott324). This shift retards antibody induction by Th2 cells against bacteria. These various ways of evasion allow bacteria to survive and proliferate in host tissues and sustain chronic infection.
Iron and nitric oxide
Bacterial cell wall components, including LPSs and PGNs, are highly resistant to degradation by mammalian enzymes and persist indefinitely in mammalian tissues. During chronic exposure, bacteria and bacterial debris accumulate in infected host tissues, sustaining chronic inflammation and slowly progressive cell damage (Refs Reference Fox272, Reference Stimpson273, Reference Fleming, Wallsmith and Rosenthal274, Reference Hauss-Wegrzyniak, Vraniak and Wenk325, Reference Lehman326). Bacteria, LPSs and PGNs have a variety of biological actions in mammals (Ref. Reference Johannsen327). They not only are inflammatory cytokine inducers and activators of complement pathways, but they also affect vascular permeability (Ref. Reference Schwab328), induce nitric oxide and free radicals, inhibit DNA synthesis, and cause apoptosis and cellular damage (Ref. Reference Heiss329).
Macrophage regulation of immune surveillance involves iron depletion (Refs Reference Weinberg and Miklossy330, Reference Griffiths331, Reference Weinberg332). Lactoferrin, which is similar in structure to transferrin, has a role in natural defence mechanisms in mammals and is upregulated in neurodegenerative disorders. Lactoferrin exerts its anti-inflammatory action by inhibiting hydroxyl radical formation. This antioxidant property prevents DNA damage (Ref. Reference Heiss329). By contrast, free iron abolishes the bactericidal effects of serum and strongly enhances infection and bacterial virulence (Refs Reference Heiss329, Reference Weinberg and Miklossy330, Reference Griffiths331, Reference Weinberg332). Iron has been shown to increase the formation of reactive oxygen intermediates, resulting in lipid peroxidation and subsequent oxidative damage of proteins and nucleic acids (Refs Reference Weinberg and Miklossy330, Reference Griffiths331, Reference Weinberg332). By affecting T-cell generation, iron also influences antigen-specific cellular responses, T-cell functions and the production of proinflammatory cytokines by macrophages (Ref. Reference Griffiths333). Iron, which has a fundamental role in infection, also accumulates in senile plaques in AD (Refs Reference Weinberg and Miklossy330, Reference Goodman334, Reference Bishop335, Reference Miller336).
Activation of macrophages and other host cells by bacteria or LPS causes inducible nitric oxide synthase (iNOS) synthesis, which in turn generates substantial amounts of nitric oxide from the amino acid l-arginine (Ref. Reference Weinberg332). Nitric oxide is a critical component in the clearance of bacterial, viral, fungal and parasitic infections (Ref. Reference Lirk, Hoffmann and Rieder337). In a mouse model, genetic disruption of iNOS was associated with a significantly higher risk of dissemination and mortality of infection (Ref. Reference Chan, Chan and Schluger338). Pathogens have also evolved a broad array of strategies to limit nitric oxide production (Refs Reference Chan, Chan and Schluger338, Reference Chakravortty and Hensel339, Reference Shiloh and Nathan340), which might contribute to their evasion from host immune responses (Ref. Reference Shiloh and Nathan340). Nitric oxide also has a central role in chronic degenerative diseases, including AD (Ref. Reference Bogdan341).
Amyloidogenesis
Amyloidogenesis is the aggregation of soluble proteins into detergent-insoluble filamentous structures about 10 nm wide and 0.1–10 µm long. These amyloid fibrils have distinct biochemical and biophysical properties, including resistance to proteinase K treatment, β-sheet structure and affinity for binding thioflavin S and Congo Red.
Chronic bacterial infections (e.g. rheumatoid arthritis, leprosy, tuberculosis, syphilis, osteomyelitis) are frequently associated with amyloid deposition. Cortical and vascular amyloid deposition in the atrophic form of general paresis, caused by a spirochete (T. pallidum), as in AD, corresponds to Aβ.
On the basis of previous observations, it has been suggested that amyloidogenic proteins might be an integral part of spirochetes and can contribute to Aβ deposition in AD (Ref. Reference Miklossy146). It was shown that the BH(Reference Terry9–Reference Kang10) peptide on a β-hairpin segment of B. burgdorferi OspA forms amyloid fibrils in vitro that are similar to those in human amyloidosis (Ref. Reference Ohnishi, Koide and Koide342). Recent observations indicate that amyloid proteins constitute a previously overlooked integral part of the cellular envelope of many bacteria (Refs Reference Otzen and Nielsen343, Reference Chapman344, Reference Larsen345, Reference Jordal346). Amyloid fibril formation not only results in toxic aggregates, but also provides biologically functional molecules (Refs Reference Otzen and Nielsen343, Reference Chapman344, Reference Wang, Hammer and Chapman347). Bacterial amyloids are involved in bacterial cell–cell interactions, in their attachment to inert solid surfaces, and in spore and biofilm formation (Ref. Reference Wang, Hammer and Chapman347). Microbial amyloids, through interaction with host proteases, also contribute to bacterial virulence, to colonisation of the host and to invasion of host cells.
Bacterial LPSs and PGNs are used worldwide to generate in vitro and in vivo experimental inflammation and amyloidosis (Ref. Reference Picken348). LPSs induce Aβ accumulation, increased APP levels and hyperphosphorylation of tau in vitro and in vivo (Refs Reference Miklossy261, Reference Hauss-Wegrzyniak and Wenk349, Reference Miklossy350, Reference Kitazawa351). Increased APP mRNA in response to LPS was also detected in the basal forebrain and hippocampus of the rat (Ref. Reference Hauss-Wegrzyniak and Wenk349). Aβ deposition occurs in the brains of rats chronically infused with LPSs (Ref. Reference Hauss-Wegrzyniak and Wenk349). In a triple-transgenic mouse model of AD, repeated challenges with LPSs were shown to exacerbate CNS inflammation and to cause increased tau phosphorylation (Ref. Reference Kitazawa351).
Genetic factors
Host responses to bacterial infections are genetically controlled. Promoter polymorphisms in the genes of proinflammatory cytokines are associated with susceptibility to infection (Ref. Reference Knight and Kwiatkowski352). Tumour necrosis factor alpha (TNF-α) is a critical mediator of host defence against infection. Polymorphisms in the gene encoding TNF-α might determine a strong cell-mediated immune response or a weak or absent cellular response, which reflects the genetic variability in cytokine production (Refs Reference Knight and Kwiatkowski352, Reference Shaw353). In the absence of cell-mediated immune responses, the microorganism can spread freely and accumulate in infected host tissues (Ref. Reference Roy354). Accordingly, in Mycobacterium leprae infection, two distinct phenotypes can be distinguished: tuberculoid leprosy and the lepromatous leprosy. In the tuberculoid or paucibacillary form, there is strong inflammatory infiltration and the number of microorganisms is low. Conversely, in the lepromatous or bacillary form, the inflammatory infiltrates are poor or absent and the number of M. leprae is high. A similar polarity in host reactions – the infiltrative form with strong cell-mediated immune responses and few spirochetes versus the atrophic form, lacking lymphoplasmocytic infiltrates but with numerous spirochetes – occurs in response to T. pallidum and B. burgdorferi infection (Refs Reference Pacheco e Silva114, Reference Pacheco e Silva115, Reference Miklossy, Duyckaerts and Litvan118, Reference Miklossy149). The influence of TNF-α polymorphism on spirochetal infections has also been demonstrated by others (Refs Reference Marangoni355).
Class II major histocompatibility genes influence host immune responses to bacterial and viral infections. The major histocompatibility complex phenotype of the antigen-presenting cell can modulate Th1-like versus Th2-like cell activity against M. leprae and other pathogens. In general, it has been acknowledged that human leukocyte antigen (HLA) DR isotypes are associated with a protective response, whereas DQ isotypes are associated with the multibacillary lepromatous form with a limited cellular response. HLA gene polymorphism is a dominant marker of susceptibility to various infections, including B. burgdorferi infection (Ref. Reference Steere, Dwyer and Winchester356). The important role of the HLA system in controlling cell-mediated responses suggests that differences in HLA haplotypes could contribute to the wide spectrum of immune responses observed in leprosy and in other infections (Ref. Reference Lagrange and Abel357). It is noteworthy that TNF-α and HLA polymorphisms, which are risk factors for infection, substantially influence the risk of AD (Refs Reference McGeer and McGeer61, Reference Collins358, Reference McCusker359, Reference Gnjec360, Reference Alves361, Reference Ballerini362).
Association between AD pathology and IL-1β expression patterns (Ref. Reference Griffin363), and between disease risk and IL-1β gene polymorphisms, has been reported (Refs Reference Bertram62, Reference Nicoll364). Genetic variation in the expression of microbial PRRs, including TLRs, and CD14 gene polymorphisms predispose to various infections and are also associated with AD (Refs Reference Minoretti365, Reference Balistreri366).
Genetic mutations in APP, PS1 and PS2 are related to the processing of APP (Ref. Reference Hardy39). Because APP has an important role in the regulation of immune system reactions and in T-cell differentiation (Refs Reference Allen16, Reference Mönning17, Reference Ledoux18), genetic defects in such genes should also result in increased susceptibility to infection.
APOE ɛ4 enhances the expression of inflammatory mediators (Refs Reference Urosevic and Martins367, Reference Licastro368) and has a modulatory function in susceptibility to infection by various bacteria, viruses and protozoa (Refs Reference Urosevic and Martins367, Reference Licastro368, Reference Corder369, Reference Itzhaki370, Reference Itzhaki and Wozniak371, Reference Bhattacharjee372). APOE genotyping of three AD cases, where B. burgdorferi was detected and cultivated from the brain, showed that two of them were APOE ɛ4 carriers. The low number of cases does not allow any conclusion for an eventual link between spirochetal infection and the APOE ɛ4 allele (Ref. Reference Miklossy149). Sixty-four per cent of AD cases with positive C. pneumoniae PCR had at least one APOE ɛ4 allele (Ref. Reference Hudson and Dolmer207). ISH and quantitative real-time PCR analyses indicated that the number of C. pneumoniae-infected cells and the bacterial load in affected brain regions of APOE ɛ4 carriers in AD were significantly higher compared with those lacking that allele (Ref. Reference Gérard200). Furthermore, HIV-1-infected subjects carrying the APOE ɛ4 allele have higher recorded levels of dementia (Ref. Reference Corder369), and the ɛ4 allele has also been shown to modulate HSV-1 infection (Refs Reference Beffert240, Reference Burgos259, Reference Miller and Federoff260, Reference Itzhaki and Wozniak371).
Outstanding research questions and conclusions
The analysis of all positive and negative data available in the literature on the association of pathogens with AD indicates a statistically significant association between various types of spirochetes, C. pneumoniae and AD. There is no significant difference between the frequency of HSV-1 in AD cases compared with age-matched controls. However, several authors found a significant difference between the frequencies of HSV-1 in APOE ɛ4 carriers and noncarriers. The occurrence of positive anti-HSV-1 IgM was reported to be a risk factor for AD (for a recent review, see Ref. Reference Itzhaki and Wozniak373).
Lesions that are similar to senile plaques, neurofibrillary tangles and neuropil threads, and granulovacuolar degeneration, accumulation of Aβ, increased APP levels and phosphorylation of tau have all been induced by exposure of mammalian neuronal and glial cells and CNS organotypic cultures to spirochetes. Aβ-positive plaques were induced by inhalation of C. pneumoniae in mice in vivo, and exposure to HSV-1 increased the Aβ level and produced tau phosphorylation in neurons in vitro and in vivo.
Through TLRs and other PRRs, pathogens or their toxic components induce gene expression and activation of proinflammatory molecules by host cells. Both the classical and alternative complement pathways are activated in AD. MAC(C5b-9), which is intended to lyse bacteria or encapsulated viruses, and activated microglia that are designed to clean up debris and foreign bacteria are both associated with cortical AD lesions.
Evasion of pathogens from host defence reactions results in sustained infection and inflammation. The microorganisms and their toxic components can be observed in affected brains, along with host immunological responses.
Spirochetes, C. pneumoniae and HSV-1 disseminate from the primary site of infection to the brain usually through systemic infection. As in syphilis, systemic infection and inflammation precede the development of dementia by years or decades. Detection of infection in its early, peripheral stage can hamper its dissemination to the CNS and prevent dementia. Consequently, peripheral infections can have a role in the initiation and progression of neurodegeneration in AD (Refs Reference Miklossy146, Reference Perry, Cunningham and Holmes374, Reference Perry, Nicoll and Holmes375, Reference Holmes and Cotterell376). Worsening of peripheral and systemic infection will deteriorate CNS involvement. One example is periodontitis, which is caused mostly by Gram-negative bacteria, including periodontal spirochetes, which represents a risk factor for AD (Refs Reference Kamer156, Reference Kamer377).
It is important to consider that several types of spirochetes and several types of pathogens can occur in AD. Coinfection of B. burgdorferi with various periodontal spirochetes (Ref. Reference Riviere, Riviere and Smith147) or with T. pallidum (Ref. Reference Blatz378) is well documented. T. pallidum frequently coinfects with other bacteria and herpes viruses in syphilis (Ref. Reference Gastinel167). In Lyme disease, coinfection of B. burgdorferi with C. pneumoniae and HSV-1, which are also associated with AD, can be observed. Coinfection by several microorganisms can accelerate the degenerative process, exacerbate CNS damage and worsen dementia.
An infectious origin of AD is in harmony with recent observations showing that Aβ belongs to the group of AMPs, which are potent, broad-spectrum bactericides targeting Gram-negative and Gram-positive bacteria, enveloped viruses, fungi and protozoans (Ref. Reference Soscia31). Aβ has the capacity to associate with lipid bilayers of bacterial cell membranes and to exert antimicrobial activity by membrane permeabilisation and by alteration of calcium homeostasis (Refs Reference Soscia31, Reference Bolintineanu379). The microtubule-binding site of tau protein also exhibits antimicrobial properties (Ref. Reference Kobayashi380).
Inflammation has a primordial role in the elimination of invading pathogens and infected host cells. If infection is eradicated, it helps the healing process. Some proinflammatory molecules, such as cytokine IL-1β, in addition to their harmful effects, have a beneficial and protective effect (Ref. Reference Shaftel381). Therefore, long-term use of anti-inflammatory drugs alone might weaken the elimination of pathogens and facilitate their evasion, survival and slowly progressive proliferation. Combined antibiotic, antiviral and anti-inflammatory therapy is suggested as the treatment of choice.
In conclusion, the data available indicate that infectious agents can initiate the degenerative process in AD, sustain chronic inflammation, and lead to progressive neuronal damage and amyloid deposition. The accumulated knowledge, views and hypotheses proposed to explain the pathogenesis of AD fit well with an infectious origin of the disease. The outcome of infection is determined by the genetic predisposition of the patient, by the virulence and biology of the infecting agent, and by various environmental factors, such as exercise, stress and nutrition.
More attention and support is needed for this emerging field of research. Infection starts long before the manifestation of dementia; therefore, an adequate treatment should start early. Because antibacterial, antiviral and anti-inflammatory therapy is available, as in syphilis, one could prevent and eradicate dementia. The effect on the suffering of patients and on the reduction of healthcare costs would be considerable.
Acknowledgements and funding
I am grateful to all those colleagues and friends who strongly supported my work on this emerging field of research during the last two decades. They all contributed, in different ways, to the realisation of this work. I thank B. Balin, A. Hudson, J. Hudson and R. Itzhaki, who have reviewed and completed data related to their field of research on C. pneumoniae and HSV-1, and R. Kraftsik for his help with the statistical analysis. This work was funded by the Prevention Alzheimer International Foundation, Switzerland. I am grateful to the peer reviewers and to the editors of the journal, who significantly contributed with their constructive advice and remarks.