


The original incentive triggering the development of the health technology assessment (HTA) system in the Czech Republic was provided by the Decision of the Czech Constitutional Court (1). By this decision, the Constitutional Court annulled a specific part of Act (2), on Public Health Insurance, particularly the provisions regarding reimbursement of medications from public health insurance funds. The Court ruled that all formal decisions on determination of price and reimbursement price must be a result of an individual administrative proceeding, that applicant has to be given a statement of reasons for the decision, and that the decision is subject to judicial review, in concordance with Council Directive 89/105/EEC (3).
Based upon this ruling of the Constitutional Court, the legislature has elaborated an entirely new regimen for the reimbursement of medicinal products with the State Institute for Drug Control (SUKL) assuming the central role in the process. These changes entered into legal force and effect on January 1, 2008. From this date on, the price and reimbursement price of medicine has been set by SUKL in formal administrative proceedings (using verifiable criteria, with a possibility of judicial review), whereas previously the out of factory price was set by the Ministry of Finance (price ceiling) and the reimbursement price and conditions by the Ministry of Health (by a Decree).
With regard to quite a brief history of the pricing and reimbursement activities of SUKL and very recently developed in-house methodology for assessing pharmacoeconomic analyses, HTA in the Czech Republic is described rather poorly in English language literature. However, the description provided by Gulacsi et al. (Reference Gulacsi, Rotar and Niewada4) in 2014 still reflects the current situation rather accurately.
HEALTHCARE SYSTEM AND ITS FINANCING IN BRIEF
The healthcare system in the Czech Republic is financed through social health insurance based on compulsory membership in a health insurance fund. The funds are quasi-public, self-governing bodies that act as payers and purchasers of health care. The chief responsibilities of the Ministry of Health include setting of the healthcare policy, supervision of the healthcare system, and drafting of healthcare legislation. The Ministry also supervises certain healthcare institutions and bodies, such as the public healthcare network and the SUKL. Regional authorities and the health insurance funds play an important role in ensuring the accessibility of health care, the former by registering healthcare providers, the latter by contracting them.
Eligible residents may freely choose their health insurance fund and healthcare providers. The health insurance funds must accept all applicants who have a legal basis for entitlement; risk selection is not permitted. The basic legal framework for the assessment of medical technologies is currently provided by Act on Public Health Insurance (2) and its implementing Decree from 2011 (5). SUKL's Pricing and Reimbursement Regulation Branch determines the price and reimbursement price and conditions, and appraises the submitted clinical and economic evaluations.
The methodology of assessment evolves with increasing requirements on the quality and complexity of the submitted documentation and increasing demands on the clinical evidence (relative efficacy, clinical effectiveness, and safety) and economic evaluation. Good quality and availability of adequate evidence for assessment and approval of reimbursement in individual authorized indications or the complete spectrum thereof is crucial. Inputs from randomized controlled trials (RCTs), indirect comparisons of trials with similar design and populations, comparisons to real-world data obtained in registries, all these diverse data resources may create inconsistencies that must be methodically and thoroughly addressed. In its activities, SUKL use in-house standard operating procedures and check lists that are publicly available (6).
ROLE OF SUKL IN PRICING AND REIMBURSEMENT OF PHARMACEUTICALS
SUKL perform appraisal of the submitted evidence, which covers the clinical- and cost-effectiveness evidence and budget impact analyses submitted by marketing authorization holders (MAHs). The opinion of SUKL is then translated into evaluation reports. MAHs and health insurance companies (i.e., parties to the proceedings) as well as expert societies of physicians are invited to present their comments and objections within a set time limit. Ultimately, SUKL decide on pricing and reimbursement of pharmaceuticals (see Figure 1).
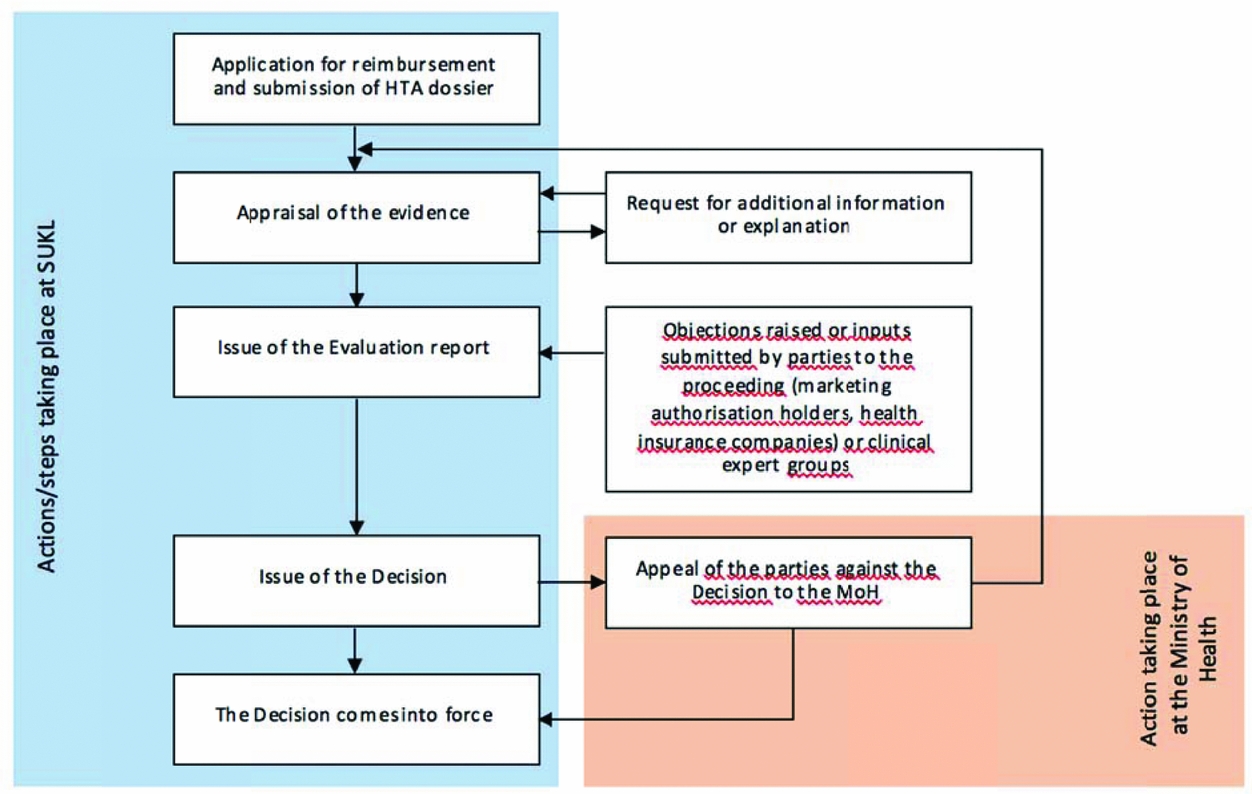
Figure 1. The scheme of the pricing and reimbursement administrative procedure. Price and reimbursement is determined or changed by means of administrative proceedings conducted by SUKL—a simplified scheme is provided below. The proceedings may be initiated upon request (as presented below) or ex officio in a “reimbursement revision” (the reason being the re-evaluation of appropriateness of reimbursement price and conditions, the course of the proceedings being similar).
The whole proceeding that takes place within SUKL is based on collaboration of small groups consisting of a lawyer, a clinical technical analyst, a pharmacoeconomic analyst, and administrative staff. Supervision is provided by the heads of individual departments (i.e., the legal, technology assessment, and administrative department). Thus, multi-level control of evaluation reports and decisions is in place to ensure quality and consistency of the evaluation process.
The estimated number of decisions issued yearly in proceedings initiated upon application is approximately 170–180 (7), approximately a quarter of the number are assessments of new medicinal products (44 decisions issued in 2015), the remaining cases being new products containing already reimbursed active substances. With regard to assessments of new products, only one of the forty-four issued decisions was negative. However, the information value of this fact is limited because MAH's reaction after a negative evaluation report can be to withdraw the application so that the proceeding is terminated without any decision being issued.
To cope with the increasing demands on quantity and quality of evaluation process, SUKL's Pricing and Reimbursement Regulation Branch recently underwent reorganization, ensuring that multidisciplinary teams can deal with each proceeding (8).
DIFFERENT LEVELS OF COMPLEXITY OF HTA PERFORMED WITHIN ADMINISTRATIVE PROCEEDINGS
HTA is at the moment implemented only for pharmaceutical products intended for out-patient care. The use of other technologies and interventions (as well as in-patient only pharmaceuticals) depends on the individual decisions of the healthcare providers and health insurance companies.
SUKL perform no horizon scanning or scoping activities, the selection of a medicinal product for outpatient care, as a new candidate for reimbursement, is made by MAH. To initiate the administrative proceeding, MAH must file an application for reimbursement for a given medicinal product together with a dossier including comprehensive documentation on clinical efficacy or effectiveness, safety, and pharmacoeconomics of the product. Upon MAH's application, an administrative proceeding is started, in course of which the submitted documentation is critically appraised and, finally, a decision is issued.
Every new medicinal product applying for permanent reimbursement must be found cost-effective. There are two major types of situations that may occur. First, complete cost-effectiveness and budget impact analyses must be provided for the new product, in addition to proofs of efficacy and safety. Or, alternatively, it must be proven that the efficacy, safety, and clinical use of the product are similar to those of other, already reimbursed products (forming a “reference group”). From the perspective of SUKL, HTA is a combination of assessment of clinical efficacy (together with effectiveness if it is available) and cost effectiveness. Medical evaluation of benefit of the assessed treatment versus the comparator represents the decisive factor for obtaining reimbursement.
Costs per quality-adjusted life-year (QALY) (or life-year gained) are not the only parameters of the assessment. However, the willingness-to-pay threshold is an integral part of the cost-effectiveness assessment. It is defined in the Czech legislation as a ratio of incremental costs and incremental benefits, which is comparable to the ratios calculated for already reimbursed therapeutic procedures at the time of their assessment. This means that the acceptability of incremental cost-utility ratio and incremental cost-effectiveness ratio (ICER) of new treatments depends on the cost-effectiveness ratios already accepted by SUKL for currently reimbursed medicines intended for a specific diagnosis. However, in the cases where no relevant comparators in the therapeutic area exist, the recommendation of the World Health Organization, that is, three times gross domestic product per capita per QALY (Reference Marseille, Larson, Kazi, Kahn and Rosen9), is currently used.
With regard to medicinal products that may be included in a “reference group,” it is not necessary to provide a complete HTA. The “reference group” is a group of pharmaceuticals that show similar effectiveness, safety profile, and clinical use and are, therefore, considered to be therapeutically interchangeable. The list of reference groups and pharmaceuticals included therein is defined by a Decree of the Ministry of Health (10) and, where appropriate, also SUKL is competent to define new groups. The Decree, which has been in force since April 2012, determines 114 groups of pharmaceuticals based on therapeutic indications (10).
In most cases, these groups associate drugs on the ATC5 level but it is not always the case. All pharmaceuticals within the same reference group have the same reimbursement price (per equipotent dose). It is also possible to set a “premium” reimbursement price in the case that a drug has higher efficacy, better safety profile, or adherence rate than the reference product, that is, the product marketed for the lowest out-of-factory price in countries included in the external referencing system. The efficacy and safety of treatment is typically assessed based on the marketing authorization study or synthesized evidence.
If a broader spectrum of evidence is available, levels of evidence are respected. The strongest evidence is represented by meta-analyses and systematic reviews, which are followed by randomized controlled trials, observational studies, nonrandomized controlled studies or follow-up studies, case control studies, or case reports, considerations based on a mechanism according to the Oxford Centre for EBM 2011. The complexity of assessment depends on the nature of the evaluated product: whether it is a product included in a reference group, new molecule launched on the market, a “highly innovative” product, or a product applying for reimbursement in an “off-label” indication.
PRINCIPLES OF SETTING THE REIMBURSEMENT CONDITIONS OF MEDICINES
Some medicinal products are reimbursed with no reimbursement conditions in place, which means that they are reimbursed in all the indications included in the “Therapeutic Indications” section in the Summary of Product Characteristic (SmPC). If a medicinal product has a condition (restriction) of reimbursement, the reimbursement from health insurance is limited to a specific indication specified in the condition, for example, to a specific disease or group of patients. In this case, if there is an extension of the therapeutic indications in the SmPC, the medicinal product is not reimbursed in this new indication.
There are two ways how to change the conditions of reimbursement: either in an individual administrative proceeding initiated upon application of an MAH or a health insurance company, or in a “reimbursement reassessment proceeding” performed regularly by SUKL in 5-year intervals. In both cases, evaluation of evidence on efficacy, effectiveness, safety, and clinical use as well as on cost-effectiveness and budget impact is necessary for the inclusion of the new indication in the reimbursement condition.
“HIGHLY INNOVATIVE MEDICINAL PRODUCT” STATUS: SPECIFIC FEATURES OF ASSESSMENT
Reimbursement of “highly innovative medicinal products” (HIMPs) may be granted for a limited period of 24 months, which may be followed by another temporary reimbursement of 12 months, that is, maximum of 36 months in total. If applying for temporary reimbursement, cost-effectiveness needs not to be proven; however, cost-effectiveness analysis is required to be submitted together with a proof of an acceptable budget impact estimate. This facilitates subsequent appraisal during the administrative proceeding dealing with permanent reimbursement when cost-effectiveness is to be proven.
In the case that an original product claims high innovation and applies for the HIMP status (many of these products are orphan medicines), it is also necessary to prove that it meets at least one of the criteria defined by the law (see Table 1). In these cases, it is also necessary to present a written commitment outlining how the applicant shall ensure continuous evaluation of treatment outcomes, limitation of budget impact, cost-effectiveness evaluation, coverage of treatment costs for the case that the period of temporary reimbursement expires.
Table 1. Criteria for Assessing the “Highly Innovative Medicinal Product” Status

Note. The criteria for granting the HIMP status take into account either improved efficacy or safety of the medicine or high unmet medical need. However, the criteria do not ensure that the new treatment brings clinically meaningful outcomes.
Oncological therapy occupies the largest share of reimbursed highly innovative medicines. The status of high innovation may disappear with market launch of new therapies, especially in those cases where the status was granted based on inexistence of reimbursed treatment alternative. Orphan medicinal products are treated just as other new molecules (there are no specific legal regulations for orphans), in most cases they fulfil the criteria of highly innovative medicinal products. Some orphan or ultra-orphan medicines may be approved despite their high ICER, with regard to high unmet medical need and low budget impact and with the approval of health insurance companies. In total, approximately 40 percent of European Medicines Agency (EMA) -registered orphan medicinal products are available and reimbursed (thirty-nine products of eighty-nine), five EMA-registered orphans are currently being evaluated (see Figure 2).
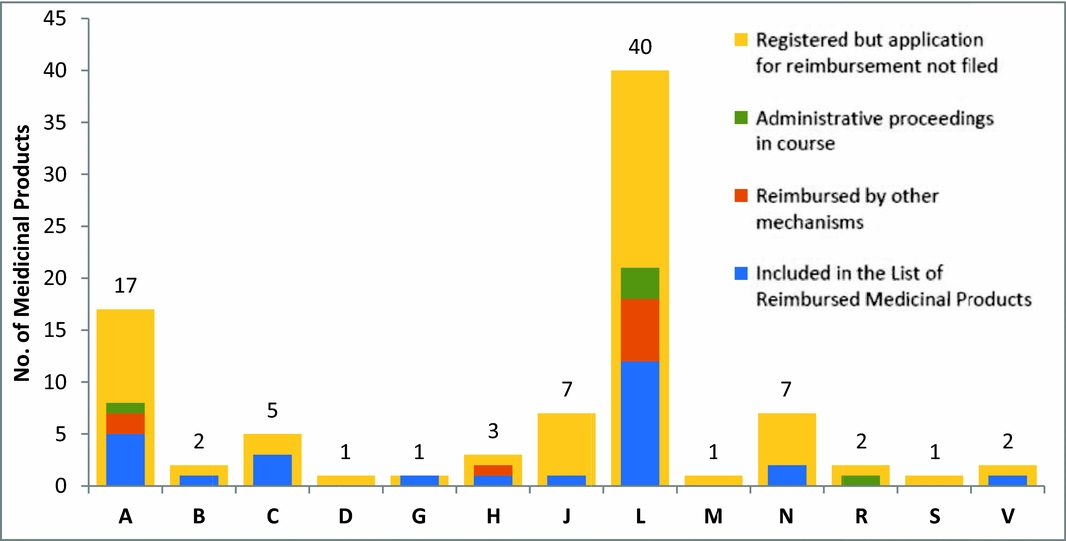
Figure 2. Representation of “orphans” in individual therapeutic groups of HIMPs, A digestive tract and metabolism; B, blood and blood-forming organs; C, cardiovascular system; D, dermatology; G, urogenital tract and sex hormones; H, systemic hormonal treatment excluding sex hormones and insulins; J, anti-infective agents for systemic use; L, cytostatic and immunomodulatory agents; M, musculoskeletal system; N, nervous system; R, respiratory system; S, sensory organs; V, other various agents. Source: List of Reimbursed Medicinal Products 10/2015 (available at: http://www.sukl.eu/sukl/list-of-reimbursed-medicinal-products-valid-as-of-1-10-2015). The graph reflects the activity of SUKL from 2008 until October 2015. Medicinal products not included in the List of Reimbursed Medicinal Products may be reimbursed by other mechanisms (they may be covered by a specific treatment program approved by the Ministry of Health or reimbursed upon individual request filed by the treating physician and approved by the health insurance company physician).
In the case of orphans, HIMPs, as well as other medicinal products, the Institute may restrict the reimbursement conditions as compared to those proposed by the applicant to a subset of patients likely to get the greatest benefit from the treatment or those pretreated with specific therapies. Otherwise, the Institute is bound by the Application and may not grant broader reimbursement conditions than those proposed by the MAH.
Pursuant to Act on Public Health Insurance, the competences and obligations of the State Institute for Drug Control include, inter alia, regular in-depth revisions of maximum prices and reimbursement prices and conditions and shortened reimbursement revisions. The latter are initiated after market launch of the first generic product, when savings exceeding € 1.11 million are anticipated, or with the aim to ensure full reimbursement of the least costly medicinal product within a reference group. Pricing and reimbursement regulation can thus be regarded as a system balancing demands on rational management of public health funds and on accessibility of good-quality medical care.
REIMBURSEMENT IN OFF-LABEL INDICATIONS
The Institute may determine a reimbursement price of a nonregistered medicinal product if its use is sufficiently substantiated by current scientific knowledge and, at the same time, if it represents the only treatment alternative or its use is cost-effective when compared with the available treatment. This also applies to nonauthorized indications of authorized medicinal products. In exceptional cases, the health insurance company may reimburse medical care that is not included in the List of reimbursed medicines, if this care represents the only option of therapy with regard to the medical condition of a specific patient. However, this reimbursement must be approved by a specialized physician (employed by the insurance company).
UNMET NEEDS OF HTA AND OBSTACLES FOR IMPLEMENTATION
With the introduction of new and effective therapeutic approaches, such as immunotherapy, the costs of treatment rapidly rise. As the novel expensive treatments usually meet the HIMP criteria, they may enter the system while not being cost-effective for the period of 2 or 3 years. Moreover, being intended for patients with nonorphan diseases, these products represent a huge burden on the public finances that might destabilize the healthcare system as the tax money flowing into the health insurance funds remains stable. More efficient tools are needed to ensure more effective regulation of growing costs of medicines, mainly those from the ATC group of “antineoplastic and immunomodulating agents”.
A variety of stakeholders (industry, payers, healthcare providers) have expressed an interest to include an HTA, in the formal or informal way, before the formal marketing authorization is granted to a medicinal product (e.g., project STAMP), but also after the formal determination of reimbursement, to manage the market entry, and the real financing (budget setting) for a particular treatment. However, such implementation is currently out of the remit of SUKL
CHALLENGES FOR THE FUTURE
Recently, SUKL has been facing some persistent obstacles that need to be constantly overcome. There is a shortage of people educated in medical statistics, research methods in medicine, or HTA, which raises demands on the time consumed by fundamental training provided within the organization. This also leads to lack of research in healthcare policy and healthcare delivery and their connection to social care. It can be seen that establishment of formal requirements on HTA in the Czech Republic has stimulated the demand for clinical and health-related economic data. Nevertheless, their accuracy and usability for the reimbursement purposes are sometimes insufficient.
Because not only the pharmaceutical products but also, for example, advanced therapies and medical devices are reimbursed from the health insurance resources, it is crucial to establish a similar methodology of HTA also for these technologies. SUKL actively participate in European activities concerning HTA so that knowledge and experience within this field is shared across HTA agencies.
CONFLICTS OF INTEREST
The authors have nothing to disclose.




 Open access
Open access