


Introduction
Eurytrema pancreaticum, also known as the pancreatic fluke, is one of the most common trematodes, living predominantly in the pancreatic and bile ducts of ruminants, such as cattle, sheep, camels and deer, and accidentally infecting humans (Sakamoto & Oikawa, Reference Sakamoto and Oikawa2007). Eurytremiasis is endemic to many countries throughout the world, causing serious losses in the livestock industry. Its symptoms include rapid weight loss, oedema, reduced performance and even death from heavy infections (Graydon et al., Reference Graydon, Carmichael, Sanchez, Weidosari and Widjayanti1992). In recent years, several studies of E. pancreaticum have focused on its epidemiology, and the diagnosis, treatment and prevention of eurytremiasis (Graydon et al., Reference Graydon, Carmichael, Sanchez, Weidosari and Widjayanti1992; Sakamoto & Oikawa, Reference Sakamoto and Oikawa2007). Despite a few molecular-level studies of E. pancreaticum (Chang et al., Reference Chang, Liu, Gao, Zheng, Zhang, Duan, Yue, Fu, Su, Gao and Wang2016; Liu et al., Reference Liu, Xu, Song, Wang and Zhu2016), little molecular research has addressed its complete ribosomal DNA sequence.
The nuclear ribosomal DNAs (rDNAs) of eukaryotes are arranged into tandem repeats. The transcriptional unit contains three genes (18S, 5.8S and 28S rRNA) with two internal transcribed spacers (ITS1 and ITS2) separating the genes and an intergenic spacer (IGS) between the transcriptional units of each repeat (Long & Dawid, Reference Long and Dawid1980). The IGS region can be further divided into two external transcribed spacers (ETSs) and a non-transcribed spacer (NTS). The two ETSs are located at the 5′-end of the 18S rDNA and the 3′-end of the 28S rDNA. The NTS is located between the two ETSs. Different rDNA regions have evolved at different rates, so they can be used as genetic markers for phylogenetic studies at different taxonomic levels (Orosová et al., Reference Orosová, Ivica, Eva and Marta2010; Yamada et al., Reference Yamada, Yoshida, Yoshida, Kuraishi, Hattori, Kai, Nagai, Sakoda, Tatara, Abe and Fukumoto2011; Dai et al., Reference Dai, Liu, Song, Lin, Yuan, Li, Huang, Liu and Zhu2012; Wang et al., Reference Wang, Gao, Zhu and Zhao2012). The IGS rDNA contains repeat sequences that contribute considerable amounts of the intraspecific and interspecific variations in parasites (Zhao et al., Reference Zhao, Blair, Li, Li, Lin, Zou, Sugiyama, Mo, Yuan, Song and Zhu2011). However, the IGS region of parasites is relatively poorly characterized (Zheng et al., Reference Zheng, Chang, Zhang, Tian, Lou, Duan, Guo, Wang and Zhu2014). Many incomplete ribosomal sequences have been reported, most of which lie between the 18S and 28S rDNAs, and form part of 28S sequences. Therefore, we determined the complete rDNA sequence of E. pancreaticum and analysed its characteristics.
Materials and methods
Parasites and DNA extraction
Adult E. pancreaticum flukes were collected from the pancreases of naturally infected beef cattle in Qiqihaer, Heilongjiang Province, China. The five adult flukes were washed thoroughly with physiological saline and identified to the species level based on their morphological features, as described previously (Olsen, Reference Olsen1974). The total genomic DNAs were extracted from the five individual adult flukes with the TIANamp Genomic DNA Kit (Tiangen, Beijing, China), according to the manufacturer's instructions, and eluted into 50 μl of double-distilled water. The DNA samples were then stored at −20°C until use.
Amplification, sequencing and assembling of complete rDNA sequences
Five pairs of primers were designed based on the corresponding rDNA sequences available in GenBank. The primer sequences and related information are listed in table 1. The polymerase chain reactions (PCRs) were performed in a total volume of 25 μl, which contained 1 μl of DNA template, 2.5 μl of 10× Ex Taq buffer, 2 μl of deoxynucleoside triphosphate (dNTP) mixture (2.5 mm each dNTP), 0.5 μl of each primer (25 mm), 0.2 μl of Ex Taq DNA polymerase (5 U/μl) and 18.3 μl of distilled water. The reactions were performed in a thermocycler under the following conditions: 95°C for 2 min (initial denaturation); followed by 35 cycles of 95°C for 1 min (denaturation), 54.6–58.8°C for 1 min (annealing) and 72°C for 1 min/kb (extension); with a final extension at 72°C for 5 min. Each amplicon was examined in a 1.0% (w/v) agarose gel, stained with ethidium bromide and photographed with transillumination. The DL2000 marker was used to estimate the sizes of the rDNA amplicons. Representative PCR products were sent to Life Technology Company (Beijing, China) for sequencing with the same primers used for their amplifications. The five complete rDNA sequences of E. pancreaticum were assembled using the DNASTAR software (Burland, Reference Burland2000).
Table 1. Primers used to amplify the complete rDNA sequence of Eurytrema pancreaticum.

F, forward primer; R, reverse primer.
Sequence analyses of complete rDNA of E. pancreaticum
The complete 18S rDNA sequence of E. pancreaticum was determined by comparing it with those of Paramphistomum cervi (GenBank accession numbers: KJ459934–KJ459938) (Zheng et al., Reference Zheng, Chang, Zhang, Tian, Lou, Duan, Guo, Wang and Zhu2014), Paragonimus kellicotti (HQ900670) and Echinostoma revolutum (GQ463130). The IGS region was determined by comparing it with those of Schistosoma japonicum (Zhao et al., Reference Zhao, Blair, Li, Li, Lin, Zou, Sugiyama, Mo, Yuan, Song and Zhu2011) and P. cervi (Zheng et al., Reference Zheng, Chang, Zhang, Tian, Lou, Duan, Guo, Wang and Zhu2014). The sequences of these rDNA regions from the individual adult flukes were aligned separately with Clustal X 1.83 (Thompson et al., Reference Thompson, Gibson, Plewniak, Jeanmougin and Higgins1997). The intraspecies sequence variations were determined with a pairwise comparison of the aligned sequences using the MegAlign procedure in DNASTAR (Burland, Reference Burland2000). The base compositions, transitions and transversions were calculated with Mega 5.0 (Tamura et al., Reference Tamura, Peterson, Peterson, Stecher, Nei and Kumar2011). The characteristics of the six regions of the E. pancreaticum rDNA were examined with REPFIND (Betley et al., Reference Betley, Frith, Graber, Choo and Deshler2002; http://cagt.bu.edu/page/REPFINDsubmit) to identify the direct repeats and EMBOSS (Rice et al., Reference Rice, Longden and Bleasby2000; http://www.bioinformatics.nl/emboss-explorer/) to find inverted repeats, using the criteria: nuclearmatches ≥ 10 bp and a mismatches ≤ 1.
Results and discussion
Complete rDNA sequences
The five complete rDNA sequences of E. pancreaticum were 8310 bp, 8309 bp, 8310 bp, 8306 bp and 8309 bp in size, similar to, or slightly shorter than, those of S. japonicum (8271–8857 bp) (Zhao et al., Reference Zhao, Blair, Li, Li, Lin, Zou, Sugiyama, Mo, Yuan, Song and Zhu2011) and P. cervi (8493–10,221 bp) (Zheng et al., Reference Zheng, Chang, Zhang, Tian, Lou, Duan, Guo, Wang and Zhu2014). The sequences have been deposited in GenBank under accession numbers KY490000–KY490004. The complete ribosomal transcription units consisted of the 18S, ITS1, 5.8S, ITS2, 28S and IGS regions, and the length of each region is shown in table 2. The A + T content was 47.71–47.85%, slightly lower than that of P. cervi (49.25–49.86%) (Zheng et al., Reference Zheng, Chang, Zhang, Tian, Lou, Duan, Guo, Wang and Zhu2014).
Table 2. Positions and lengths of the ribosomal gene sequences of five specimens of Eurytrema pancreaticum and the published E. pancreaticum sequences.
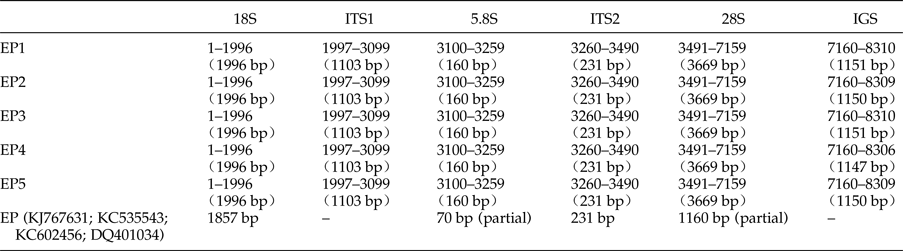
18S, 5.8S and ITS2 rDNA sequences
The length range of the 18S rDNA sequences of trematodes is usually 1836–1994 bp, whereas the length for all 18S rDNA sequences in the five pancreatic flukes analysed here was 1996 bp, which is longer than the corresponding E. pancreaticum sequence available in GenBank (KJ767631). The intraspecific variations in 18S rDNA were 0–0.2%, which are consistent with those of P. cervi. The A + T contents of 18S rDNA sequences were 49.00–49.10%, which lie within the ranges in the suborders Plagiorchiata (48.18–49.23%) and Echinostomata (48.91–49.42%) (Zheng et al., Reference Zheng, Chang, Zhang, Tian, Lou, Duan, Guo, Wang and Zhu2014), but lower than those in the suborder Strigeata (50.05–51.09%) (Zhao et al., Reference Zhao, Blair, Li, Li, Lin, Zou, Sugiyama, Mo, Yuan, Song and Zhu2011).
The previously published 5.8S sequence of E. pancreaticum in GenBank (KJ767631) is only 70 bp in length, and the length of 5.8S rDNA sequences have slight differences in parasites, with a range of 150–176 bp (Lin et al., Reference Lin, Chen, Weng, Wu, Zou, Li, Song and Zhu2005; Zhao et al., Reference Zhao, Jiang, Dong and Nie2012); even so, there are individual exceptions – the length of the 5.8S rDNA sequences of Rhipicephalus microplus (JX974346) is 1151 bp, which is the longest reported. However, the length of the 5.8S rDNA sequences of E. pancreaticum determined in this study was 160 bp, which lies within the range given above, and is the same as those of Clonorchis sinensis, E. revolutum and Echinostoma caproni. There were no intraspecific variations within the E. pancreaticum 5.8S rDNA sequence in the present study. The 5.8S rDNA is highly conserved throughout parasites. For example, a previous study found 98% similarity among the 5.8S sequences of three Moniezia species (Maiko et al., Reference Maiko, Mikiko and Tadashi2015). In the present study, the A + T content of the 5.8S rDNA sequences of E. pancreaticum was 46.25%, which is similar to that of the majority of flukes, including Dicrocoelium dendriticum (KC774524) (46.58%) and E. revolutum (U58102) (46.88%).
The length range of the ITS2 rDNA sequences of trematodes is 234–432 bp, whereas that of invertebrates is 100–1300 bp (Odorico & Miller, Reference Odorico and Miller1997; Harris & Crandall, Reference Harris and Crandall2000). Encephalitozoon cuniculi is a special case in that it lacks the ITS2 rDNA sequence (Peyretaillade et al., Reference Peyretaillade, Biderre, Peyret, Duffieux, Méténier, Gouy, Michot and Vivarès1998). In the present study, the length of ITS2 of E. pancreaticum was 231 bp, which is the shortest among those reported for flukes. The ITS2 rDNA sequences of the five samples of E. pancreaticum in this study were identical to the published E. pancreaticum sequence (KC535543). The A + T content of the ITS2 rDNA sequences was 46.75%, which is lower than that of D. dendriticum (KC774524) (52.14%), in the same family.
ITS1 and 28S rDNA sequences
Previously, data have shown that the length range of the ITS1 rDNA sequences of trematodes is 384–1428 bp (Zheng et al., Reference Zheng, Chang, Zhang, Tian, Lou, Duan, Guo, Wang and Zhu2014). In the present study, in the five samples investigated, the ITS1 rDNA sequence was 1103 bp, which is shorter than that of Trichobilharzia regenti (HM439497.1), but longer than those of other flukes (Karamian et al., Reference Karamian, Aldhoun, Maraghi, Hatam, Farhangmehr and Sadjjadi2011). The intraspecific variation within E. pancreaticum was 0–0.5%. The A + T content of ITS1 in our samples was 45.97–46.15%, which is higher than that in P. kellicotti (HQ900670) or C. sinensis (JQ048601), but lower than that in other trematodes.
We identified nine types of repeat sequence (types A–I) in the ITS1 of E. pancreaticum, with the following characteristics: four copies of an 11-nt complete direct repeat, A, located at 71 nt upstream of the ITS1 sequence; three copies of a 10-nt complete direct repeat, B, separated by 1 nt, which occurred after repeat A; three copies of a 13-nt nearly complete direct repeat, C, separated by 1 nt, which occurred after repeat B; four copies of a 14-nt complete direct repeat, D; four copies of a 12-nt direct repeat, E; four copies of a 19-nt direct repeat, F; four copies of an 11-nt direct repeat, G; four copies of a 19-nt direct repeat, H; and two copies of a 14-nt direct repeat, I, which contained repeat F and overlapped the third repeat B by 4 nt (fig. 1). In this study, the repeat of ITS sequence was shorter than that previously reported in P. cervi (Zheng et al., Reference Zheng, Chang, Zhang, Tian, Lou, Duan, Guo, Wang and Zhu2014). Previous studies have shown that ITS1 rDNA is less conserved than ITS2 rDNA (Luton et al., Reference Luton, Walker and Blair1992), which may be attributable to the numbers and types of repeats present (Zheng et al., Reference Zheng, Chang, Zhang, Tian, Lou, Duan, Guo, Wang and Zhu2014). Long and short repeats causing size variations have been found across a range of helminths, including trematodes (Herwerden et al., Reference Herwerden, Blair and Agatsuma1998, Reference Herwerden, Blair and Agatsuma1999; Warberg et al., Reference Warberg, Jensen and Frydenberg2005), cestodes (Bowles et al., Reference Bowles, Blair and McManus1995) and nematodes (Subbotin et al., Reference Subbotin, Deimi, Zheng and Chizhov2011), but we detected no length variations in this study.
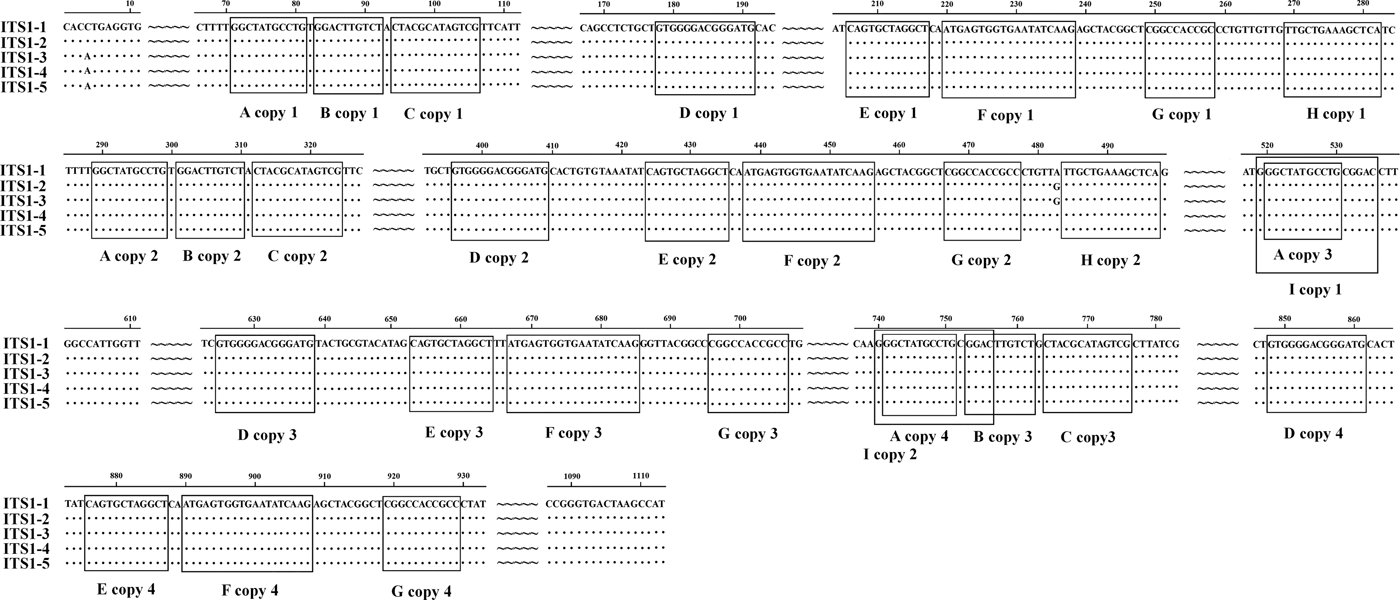
Fig. 1. Alignment of the ITS1 rDNA region of the five samples of Eurytrema pancreaticum.
The previously published 28S rDNA sequence of E. pancreaticum is a partial sequence, only 1160 bp in length (KC602456), whereas the complete 28S sequence amplified in this study had a length of 3669 bp, shorter than those of other flukes, such as S. japonicum (3897 bp) and P. cervi (4186 bp) (Zhao et al., Reference Zhao, Blair, Li, Li, Lin, Zou, Sugiyama, Mo, Yuan, Song and Zhu2011; Zheng et al., Reference Zheng, Chang, Zhang, Tian, Lou, Duan, Guo, Wang and Zhu2014). This is the first report of the complete 28S rDNA of E. pancreaticum. The intraspecific variations within E. pancreaticum 28S rDNA were 0–0.2%, which is lower than variations in that of P. cervi (0–0.5%). The A + T content was 47.23–47.32%, which is higher than that of P. cervi (45.86%) (Zheng et al., Reference Zheng, Chang, Zhang, Tian, Lou, Duan, Guo, Wang and Zhu2014), but lower than that of S. japonicum (Z46504.4) (50.24%). The 28S rDNA sequences had only two types of repeats (J and K), which were both 11 nt long and occurred in two copies (fig. 2).
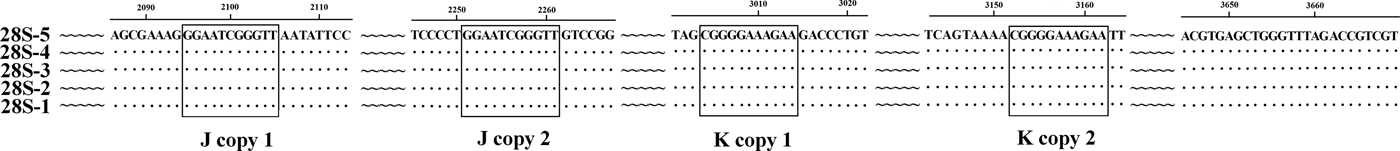
Fig. 2. Alignment of the 28S rDNA sequences of the five individual Eurytrema pancreaticum specimens.
Analyses of IGS rDNA sequences
The length variation in the published IGS sequences of trematodes ranges from 527 to 3035 bp, whereas the IGS of the five E. pancreaticum specimens analysed in this study ranged from 1147 to 1151 bp in length. This length variation of only 4 bp differs markedly from the previously reported length variation in P. cervi (577–2305 bp) (Zheng et al., Reference Zheng, Chang, Zhang, Tian, Lou, Duan, Guo, Wang and Zhu2014), but is similar to that reported previously for S. j aponicum, in which the length range in samples from different sites were 1252–1838 bp, although there was no variation among samples from the same sites (Zhao et al., Reference Zhao, Blair, Li, Li, Lin, Zou, Sugiyama, Mo, Yuan, Song and Zhu2011).
In the present study, the intraspecific variations in the IGS rDNA sequences of E. pancreaticum were 2.9–20.2%, higher than those in S. japonicum (0–2.3%) (Zhao et al., Reference Zhao, Blair, Li, Li, Lin, Zou, Sugiyama, Mo, Yuan, Song and Zhu2011), but similar to the results reported for P. cervi (0.2–46.3%) (Zheng et al., Reference Zheng, Chang, Zhang, Tian, Lou, Duan, Guo, Wang and Zhu2014). The IGS region usually contains some repeat sequences (Zhao et al., Reference Zhao, Blair, Li, Li, Lin, Zou, Sugiyama, Mo, Yuan, Song and Zhu2011; Zheng et al., Reference Zheng, Chang, Zhang, Tian, Lou, Duan, Guo, Wang and Zhu2014), but no repeat sequence was found in this study. The A + T content in E. pancreaticum was 48.74–49.83%, which is lower than that in other trematodes, including P. cervi (49.74–25.02%) (Zheng et al., Reference Zheng, Chang, Zhang, Tian, Lou, Duan, Guo, Wang and Zhu2014) and S. japonicum (56.79–58.29%) (Zhao et al., Reference Zhao, Blair, Li, Li, Lin, Zou, Sugiyama, Mo, Yuan, Song and Zhu2011).
Phylogenetic relationships
The 18S rRNA sequence is useful for studying the phylogeny of members of the subclass Digenea (Otranto et al., Reference Otranto, Rehbein, Weigl, Cantacessi, Parisi, Lia and Olson2007). Therefore, the phylogenetic position of E. pancreaticum was determined using 18S rRNA sequences. Using maximum parsimony (MP), Bayesian inference (BI) and maximum likelihood (ML) analyses, the phylogenetic relationships among members of class Trematoda were determined based on 18S rDNA sequences available in GenBank, with no gaps at either end, and with Schistosoma turkestanicum (AF442499) as the outgroup. Three trees all placed E. pancreaticum within the family Dicrocoeliidae, as shown in fig. 3. Two main clades were observed. All the trematodes in suborders Opisthorchiata and Plagiorchiata clustered together in the larger clade, and members of the suborder Echinostomata clustered alone in the other clade, in accordance with the morphological classification. In the clade including Opisthorchiata and Plagiorchiata, each suborder formed an independent cluster. The five complete 18S rDNA sequences of E. pancreaticum determined in this study (EP1, EP2, EP3, EP4 and EP5) and the E. pancreaticum 18S rDNA sequences published by the National Center for Biotechnology Information (NCBI) all clustered with Plagiorchiata. It can be seen from the tree that E. pancreaticum is more closely related to Opisthorchiata. These results are consistent with the traditional morphological classification.

Fig. 3. Phylogenetic relationships of E. pancreaticum and other trematodes constructed from 18S rDNA sequences using MP/ML/BI methods.
In conclusion, in this study we determined and characterized the complete rDNA sequences of five individual E. pancreaticum specimens for the first time. The 18S, ITS1, 5.8S, ITS2 and 28S rDNA regions were all quite strongly conserved, with no length variations, and the length variation in the IGS rDNA region was only 4 bp. A phylogenetic analysis based on the 18S rDNA sequences showed that E. pancreaticum, within the family Dicrocoeliidae of Plagiorchiata, is closely related to the suborder Opisthorchiata. These results provide information upon which to base future studies of the rDNA sequences of the Dicrocoeliidae trematodes.
Financial support
This work was supported, in part, by the Scientific Research Fund of Heilongjiang Provincial Science and Technology Department (GZ13B001), grants from the General Bureau of Land Reclamation of Heilongjiang Province (HNK125B-11-4) and Heilongjiang Bayi Agricultural University Graduate Innovative Research Project (YJSCX2016-Y17).
Conflict of interest
None.
Ethical standards
Flukes were collected from the pancreases of naturally infected beef cattle in accordance with the Animal Ethics Procedures and Guidelines of the People's Republic of China. The performance of this study was strictly according to the recommendations of the Guide for the Care and Use of Laboratory Animals of the Ministry of Health, China, and our protocol was reviewed and approved by the Research Ethics Committee of Heilongjiang Bayi Agricultural University.





