


Introduction
Monogenea, a group of largely ectoparasitic members of the flatworm phylum Platyhelminthes, mainly found on skin or gills of fish, is composed of two major subgroups, Polyopisthocotylea and Monopisthocotylea, distinguished by the morphology of attachment organs (Justine, Reference Justine1998). Sometimes an alternative nomenclature is also used: Polyonchoinea and Heteronchoinea (including Oligonchoinea and Polystomatoinea) (Boeger & Kritsky, Reference Boeger, Kritsky, Littlewood and Bray2001). Although some studies use the family name Ancyrocephalidae (Bychowsky & Nagibina, Reference Bychowsky and Nagibina1978; Lebedev, Reference Lebedev1988), it was shown to be a junior synonym for Dactylogyridae, based on both morphological characters (Kritsky & Boeger, Reference Kritsky and Boeger1989) and molecular data (Šimková et al., Reference Šimková, Plaisance, Matějusová, Morand and Verneau2003). With more than 900 nominal species, Dactylogyrus (Dactylogyrinae subfamily) is the largest helminth genus, with a globally widespread distribution (Gibson et al., Reference Gibson, Timofeeva and Gerasev1996). They are highly host-specific, commonly infecting only one host species or several congeneric hosts, but mainly cyprinids (Šimková et al., Reference Šimková, Morand, Jobet, Gelnar and Verneau2004). Dactylogyrus lamellatus (Achmerov, 1952) is a parasite commonly found on the gills of grass carp (Ctenopharyngodon idella Valenciennes, 1844). As in other monogeneans, its single-host life cycle can be completed easily in a closed system (Schäperclaus, Reference Schäperclaus1991), and it can cause high mortality in cultured grass carp fry and fingerlings (Shamsi et al., Reference Shamsi, Jalali and Aghazadeh Meshgi2009). Establishment of D. lamellatus parasites on the gill lamellae gives rise to local and general lesions (Molnár, Reference Molnár1971), and may incur ensuing secondary bacterial, viral and fungal infections (Thoney & Hargis, Reference Thoney and Hargis1991).
Previously, single molecular markers, such as partial mitochondrial rrnS, cox1 and nad2 genes, 18S and 28S rDNA genes, and internal transcribed spacer (ITS 1 and 2) of the rRNA gene, have been used to study the Dactylogyridae family. These studies focused mainly on the phylogeography (Wang et al., Reference Wang, Yan, Brown, Shaharom-Harrison, Shi and Yang2014), species identification (Borji et al., Reference Borji, Naghibi, Nasiri and Ahmadi2012), co-speciation in host–parasite assemblages (Šimková et al., Reference Šimková, Morand, Jobet, Gelnar and Verneau2004) and phylogeny of the Dactylogyridae (Šimková et al., Reference Šimková, Matějusová and Cunningham2006). Regarding the phylogeny, poly- and/or paraphyly of the ‘catch-all’ Ancyrocephalidae family (or Ancyrocephalinae subfamily) have been widely discussed (Šimková et al., Reference Šimková, Plaisance, Matějusová, Morand and Verneau2003, Reference Šimková, Matějusová and Cunningham2006; Mendoza-Palmero et al., Reference Mendoza-Palmero, Blasco-Costa and Scholz2015). As such, single gene sequences may not provide sufficient resolution. Thus, Delsuc et al. (Reference Delsuc, Tsagkogeorga, Lartillot and Philippe2008) have argued that 18S rRNA (and other single molecular markers) have a limited resolving power for phylogeny, and suggested phylogenomics as a much more powerful tool. Huyse et al. (Reference Huyse, Buchmann and Littlewood2008) have also reported that both the V4 hypervariable region of 18S and the ITS rRNA region did not provide sufficient phylogenetic resolution to distinguish between Gyrodactylus thymalli (Zitnan, 1960) and Gyrodactylus salaris (Malmberg, 1957), so they developed a mitogenomic approach for the identification of Gyrodactylus species and strains. Nevertheless, within the Dactylogyridae family, the complete mitogenomic sequence is currently available only for one representative of the Ancyrocephalinae subfamily, whereas such sequences remain unavailable for the entire large Dactylogyrinae subfamily.
Mitogenomes have several useful characteristics: haploidy, relatively high mutation rates, a lack of recombination and a rapidly increasing set of available orthologous sequences (Gissi et al., Reference Gissi, Iannelli and Pesole2008), so they have been widely used as genetic markers in population genetics (Shao & Barker, Reference Shao and Barker2007), phylogenetics (Park et al., Reference Park, Kim, Kang, Kim, Eom and Littlewood2007; Perkins et al., Reference Perkins, Donnellan, Bertozzi and Whittington2010) and diagnostics (Huyse et al., Reference Huyse, Buchmann and Littlewood2008). Notably, based on the phylogenetic analysis of the six available monogenean mitogenomes and all other published platyhelminth mitogenomes, Perkins et al. (Reference Perkins, Donnellan, Bertozzi and Whittington2010) rejected the Cercomeromorphae theory (an assertion that Monogenea is more closely related to Cestoda than to Trematoda) and found that Monogenea is paraphyletic.
Comparisons of mitochondrial gene arrangements are also emerging as a powerful tool for investigating phylogeny and systematics (Boore & Brown, Reference Boore and Brown1998). They might be particularly useful for resolving the phylogeny of Platyhelminthes, which are undergoing a very fast nucleotide substitution rate (Lavrov & Lang, Reference Lavrov and Lang2005), which can lead to mutational saturation, whereas the much slower rate of gene rearrangements preserves the phylogenetic signal for much longer periods of time. Indeed, mitochondrial gene order has proven useful for inferring phylogenetic relationships in certain groups of Platyhelminthes, including the relationships between major lineages of monogeneans, trematodes and cestodes (Park et al., Reference Park, Kim, Kang, Kim, Eom and Littlewood2007), African and Asian schistosomes (Huyse et al., Reference Huyse, Plaisance, Webster, Mo, Bakke, Bachmann and Littlewood2007), as well as monopisthocotyleans and polyopisthocotyleans (Zhang et al., Reference Zhang, Wu, Xie and Li2012).
So far, 16 complete monogenean mitogenome sequences have been published, including 13 monopisthocotylid species (9 Gyrodactylidae, 3 Capsalidae and 1 Dactylogyridae) and 3 polyopisthocotylid species (2 Microcotylidae and 1 Chauhaneidae). In the present study, we have sequenced and characterized the first mitogenome of any representative of the Dactylogyrinae subfamily, D. lamellatus. Furthermore, we have used all 36 genes to infer its phylogenetic relationships with the remaining 16 monogeneans for which a mitogenome sequence is available.
Materials and methods
Specimen collection and DNA extraction
Monogeneans were collected from the gills of grass carp specimens obtained from an earthen pond in Wuhan, Hubei Province, China. Dactylogyrus lamellatus was identified morphologically by the hard parts of the haptor (anchors, dorsal and ventral connective bars, marginal hooks) and reproductive organs (male copulatory organ and vaginal armament) (Gusev, Reference Gusev and Bauer1985), under a stereomicroscope and a light microscope. The parasites were preserved in 99% ethanol and stored at 4°C. The total genomic DNA of about 120 individual parasites was extracted from the entire parasites using TIANamp Micro DNA Kit (Tiangen Biotech, Beijing, China) according to the manufacturer's instructions, and stored at −20°C. Taxonomic identity of the specimen was further verified by amplifying a fragment of 18S rDNA and the complete ITS1 region using the upstream primer S1 (5′-ATTCCGATAACGAACGAGACT-3′) and downstream primer H7 (5′-GCTGCGTTCTTCATCGATACTCG-3′) (Sinnappah et al., Reference Sinnappah, Lim, Rohde, Tinsley, Combes and Verneau2001).
PCR and DNA sequencing
Partial sequences of cox1, cob, rrnS, nad5, nad1, cox2, nad4 and cox3 genes were initially amplified via polymerase chain reaction (PCR) using eight primer pairs (see supplementary table S1). Based on these fragments, we have designed specific primers for subsequent PCR amplification (supplementary table S1). PCR reactions were conducted in a 20-μl reaction mixture, containing 7.4 μl double-distilled water (dd H2O), 10 μl 2 × PCR buffer (Mg2+, dNTP plus; Takara, Dalian, China), 0.6 μl of each primer, 0.4 μl rTaq polymerase (250 U, Takara) and 1 μl DNA template. Amplification was performed under the following conditions: initial denaturation at 98°C for 2 min; followed by 40 cycles at 98°C for 10 s, 48–60°C for 15&h;s, 68°C for 1 min/kb; and a final extension at 68°C for 10 min. PCR products were sequenced bidirectionally at Sangon Company (Shanghai, China) using the primer-walking strategy.
Sequence analyses
The complete mitogenomic sequence of D. lamellatus was assembled manually and aligned against the mitogenomic sequences of other published monogeneans using the program MAFFT 7.149 (Katoh & Standley, Reference Katoh and Standley2013) to determine approximately the boundaries of genes. Protein-coding genes were inferred with the help of BLAST and ORF Finder tools (both available from the National Center for Biotechnology Information (NCBI)), employing the echinoderm mitochondrial genetic code (Codon table 9), and translated into amino acid sequences in MEGA 5 (Tamura et al., Reference Tamura, Peterson, Peterson, Stecher, Nei and Kumar2011). A majority of the tRNAs were identified using tRNAscan-SE web tool (Lowe & Eddy, Reference Lowe and Eddy1997), and the rest were found by visual comparison with other monopisthocotylids. rrnL and rrnS were found by alignment with other published monopisthocotylean mitogenomes, and their ends were assumed to extend to the boundaries of their flanking genes. Their secondary structures were predicted by Mfold software (Zuker, Reference Zuker2003). Tandem Repeats Finder (Benson, Reference Benson1999) was used to identify tandem repeats in the non-coding region (NCRs), and their secondary structures were predicted by Mfold software. Base composition, amino acid composition of protein-encoding genes (PCGs) and codon usage were computed with MEGA 5. Rearrangement events in the mitogenomes and pairwise comparisons of gene orders of seven monogeneans were calculated with the CREx program (Bernt et al., Reference Bernt, Merkle, Ramsch, Fritzsch, Perseke, Bernhard, Schlegel, Stadler and Middendorf2007) utilizing the breakpoint dissimilarity measurement. Due to the limitations of the program, the taxa with multiple NCRs were removed from the CREx analysis; these included: Aglaiogyrodactylus forficulatus (Kritsky et al. 2007), Gyrodactylus kobayashii (Hukuda, 1940), G. gurleyi (Price, 1937), G. salaris, G. thymalli, G. derjavinoides (Malmberg et al., 2007), G. brachymystacis (Ergens, 1978), G. parvae (You, Easy & Cone, 2008), Benedenia hoshinai (Ogawa, 1984) and B. seriolae (Yamaguti, 1934). Linear comparison of the 17 studied monogenean mitogenomes was visualized using EasyFig2.2 (Sullivan et al., Reference Sullivan, Petty and Beatson2011), with the E-value threshold set to 0.001 for BLASTn. Non-synonymous (dN)/synonymous (dS) mutation rates among the 12 PCGs of D. lamellatus and the only remaining Dactylogyridae species with available mitogenomic sequence, Tetrancistrum nebulosi (Young, 1967), were calculated with KaKs_Calculator (Zhang et al., Reference Zhang, Li, Zhao, Wang, Wong and Yu2006) using a modified Yang–Nielsen algorithm.
Phylogenetic analyses
Phylogenetic analyses were undertaken on the newly sequenced mitogenome of D. lamellatus and the 16 monogenean genomes available in GenBank (see supplementary table S4). Two Tricladida (order) species, Crenobia alpina (Dana, 1766) (KP208776) and Obama sp. MAP-2014 (NC_026978), were used as outgroups. A Fasta file with the nucleotide sequences for all 36 genes (12 PCGs, 2 rRNAs and 22 tRNAs) was extracted from the GenBank files using a home-made GUI-based program: MitoTool (https://github.com/dongzhang0725/MitoTool). This program was also used to generate the *.sqn file (for GenBank submission) by parsing the Word document's comment box, yielding the original *.csv format files for table 1 and supplementary tables S2, S3 and S4. All the genes were aligned in batches with MAFFT integrated in another home-made GUI-based program, BioSuite (https://github.com/dongzhang0725/BioSuite), wherein codon-alignment mode was used for the 12 PCGs, and normal-alignment mode for the remaining genes (rRNAs and tRNAs). BioSuite was then used to concatenate these alignments into a single alignment and generate phylip and nexus format files for the phylogenetic analyses, conducted using maximum likelihood (ML) and Bayesian inference (BI) methods. The selection of the most appropriate evolutionary models for the dataset was carried out using ModelGenerator v0.8527 (Keane et al., Reference Keane, Creevey, Pentony, Naughton and Mclnerney2006). Based on the Akaike information criterion, GTR + I+G was chosen as the optimal model of nucleotide evolution. ML analysis was performed by RaxML GUI (Silvestro & Michalak, Reference Silvestro and Michalak2012) using the ML + rapid bootstrap (BP) algorithm with 1000 replicates. BI analysis was performed in MrBayes 3.2.1 (Ronquist et al., Reference Ronquist, Teslenko, van der Mark, Ayres, Darling, Höhna, Larget, Liu, Suchard and Huelsenbeck2012) with default settings, and 1 × 107 metropolis-coupled MCMC generations.
Results and discussion
Genome organization and base composition
The circular mitogenome of D. lamellatus was 15,187 bp in length (GenBank accession number: KR871673), which is close to the longest monogenean mitogenome (15,527 bp) described so far – Polylabris halichoeres (Wang & Yang, 1998) (supplementary table S4). Apart from lacking the Atp8 gene, which is common in flatworms (Le et al., Reference Le, Blair and McManus2002), the mitogenome contained the standard 36 genes: 22 tRNA genes, 2 rRNA genes and 12 protein-encoding genes (PCGs), as well as a major NCR (fig. 1). All genes were transcribed from the same strand. Similar to other monogeneans (supplementary table S4), it had a high A + T content (70.6%) (supplementary table S2). Nine overlapping regions were found in the genome (table 1), indicating that it is highly compacted, which is typical for flatworm mitogenomes. The overlap between nad4L and nad4, although variable in length, is common in flatworm mtDNAs, with the exception of two Benedenia species, B. hoshinai and B. seriolae, whose nad4L and nad4 genes are separated by a short NCR (Perkins et al., Reference Perkins, Donnellan, Bertozzi and Whittington2010).

Fig. 1. Map of the complete mitochondrial genome of Dactylogyrus lamellatus. PCGs are red, rRNAs green and tRNAs blue. tRNA genes are labelled with the one-letter amino acid code, where L1 = trnL1 (uag), L2 = trnL2 (uaa), S1 = trnS1 (gcu) and S2 = trnS2 (uga). NCR (grey) is the main non-coding region.
Table 1. The annotated mitochondrial genome of Dactylogyrus lamellatus.
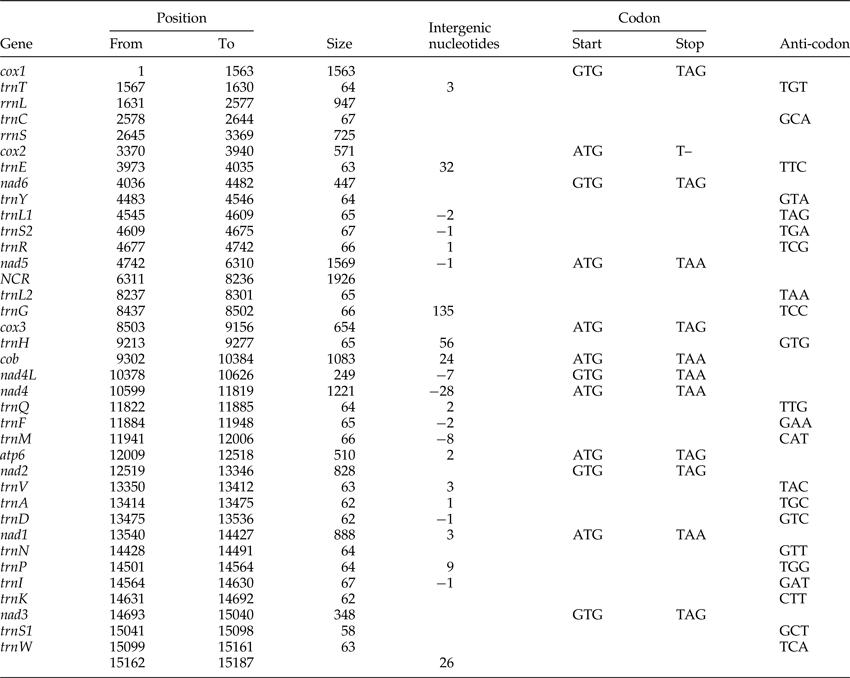
Protein-coding genes and codon usage
The total length of the concatenated 12 protein-coding genes was 9931 bp, with an average A + T content of 68.9%, varying from 63.4% (cox2) to 74.5% (nad6) (supplementary table S2). The most frequent start codon was ATG (for seven PCGs), followed by GTG (five genes). Among the terminal codons, six were TAG, five were TAA, and cox2 had an abbreviated stop codon (T–), which seems to be exclusive to D. lamellatus among the monogenean genomes published to date (supplementary table S3). Codon usage, relative synonymous codon usage (RSCU) and codon family proportion (corresponding to the amino acid usage) of the two dactylogyrids (D. lamellatus and T. nebulosi) are presented in fig. 2. Leucine (15.52%), serine (10.64%) and phenylalanine (10.43%) were the most frequent amino acids in the PCGs of D. lamellatus, while glutamine (1.24%), arginine (1.52%) and lysine (1.61%) were relatively scarce (fig. 2). The most frequent codons were TTT (phenylalanine, 10.00%) and TTA (leucine, 9.00%), whereas the CGC codon for arginine was absent. Such a preference for codon and amino acid usage (as in D. lamellatus) was also found in other Gyrodactylids (Huyse et al., Reference Huyse, Plaisance, Webster, Mo, Bakke, Bachmann and Littlewood2007, 2008; Plaisance et al., Reference Plaisance, Huyse, Littlewood, Bakke and Bachmann2007; Ye et al., Reference Ye, King, Cone and You2014; Bachmann et al., Reference Bachmann, Fromm, Patella de Azambuja and Boeger2016), capsalids (Perkins et al., Reference Perkins, Donnellan, Bertozzi and Whittington2010; Zhang et al., Reference Zhang, Wu, Li, Zhao, Xie and Li2014b) and polyopisthocotylids (Zhang et al., Reference Zhang, Wu, Xie and Li2012). From fig. 2, one cannot fail to observe that the codons ending in A or T were predominant, which corresponds to the high A + T content of the third coding position of all PCGs in D. lamellatus (76.2%) and T. nebulosi (69.1%) (supplementary table S2).

Fig. 2. Relative Synonymous Codon Usage (RSCU) of the complete mitochondrial genome of Dactylogyrus lamellatus and Tetrancistrum nebulosi. Codon families are labelled on the x-axis. Values on the top of the bars refer to amino acid usage.
The ratios of non-synonymous (dN) to synonymous (dS) substitutions (ω) for all 12 PCGs of D. lamellatus versus T. nebulosi ranged from 0.03 to 0.26 (supplementary fig. S1). All of the PCGs were under negative (purifying) selection (dN/dS < 1), suggesting the existence of functional constraints affecting the evolution of these genes. Among them, functional constraints on nad4L, nad2, nad6 and atp6 genes were the most relaxed, which was also detected in previous monogenean mitogenomic studies (Huyse et al., Reference Huyse, Buchmann and Littlewood2008; Zhang et al., Reference Zhang, Wu, Xie and Li2012; Ye et al., Reference Ye, King, Cone and You2014). On the contrary, the cox1 gene (ω = 0.03 in this study) was repeatedly found to evolve under strong selective pressure (Huyse et al., Reference Huyse, Buchmann and Littlewood2008; Zhang et al., Reference Zhang, Wu, Xie and Li2012, Reference Zhang, Wu, Li, Zhao, Xie and Li2014b; Ye et al., Reference Ye, King, Cone and You2014).
Transfer and ribosomal RNA genes
All 22 commonly found tRNAs were present in the mitogenome of D. lamellatus, ranging from 58 bp (trnS1 (gcu)) to 67 bp (trnC (gca), trnS2 (uga) and trnI (gau)) in size, and adding up to 1412 bp in total concatenated length (supplementary table S2). All of the tRNA sequences could be folded into the conventional cloverleaf structure, including an amino-acyl stem of seven nucleotide pairs (ntp), a dihydrouridine (DHU)-stem of 3–4 ntp with a 4–9 nt loop, an anticodon stem of 5 ntp with a loop of 7 nt, and a TΨC stem of 3–6 ntp with a loop of 3–6 nt (Park et al., Reference Park, Kim, Kang, Kim, Eom and Littlewood2007). trnS1 (gcu), which lacked the DHU arms, was an exception. rrnL and rrnS were 947 and 725 bp in size, with 69.8 and 66.3% A + T content, respectively (supplementary table S2). They were separated only by trnC (gca), whereas trnT (ugu) was found between rrnL and cox1, which is the standard arrangement for monopisthocotylids (Huyse et al., Reference Huyse, Plaisance, Webster, Mo, Bakke, Bachmann and Littlewood2007, Reference Huyse, Buchmann and Littlewood2008; Ye et al., Reference Ye, King, Cone and You2014; Zhang et al., Reference Zhang, Wu, Li, Xie and Li2014a, Reference Zhang, Wu, Li, Zhao, Xie and Lib) (fig. 1). Benedenia seriolae was an exception again, with a positional change of trnT (ugu) (Perkins et al., Reference Perkins, Donnellan, Bertozzi and Whittington2010) (fig. 3B). Predicted secondary structures of the two ribosomal RNA genes are displayed in supplementary fig. S2.

Fig. 3. (A) Phylogeny of the five families inferred from almost complete genomic datasets (36 genes: 12 PCGs, 2 rRNAs and 22 tRNAs), using two Tricladida species as outgroups. Scale bar represents the estimated number of substitutions per site. Bootstrap/posterior probability support values of ML/BI analysis are shown above the nodes. (B) Linear comparison of monogenean genomes (corresponding to tip labels in (A)). The more similar the sequence, the darker the grey blocks between them. tRNAs are coloured blue; protein genes, red; rRNAs, green; and the major NCR, grey.
Non-coding regions
A total of 13 short intergenic regions (1–135 bp) were interspersed within the mitogenome, adding up to a total of 297 bp (table 1). The major non-coding region (NCR), 1926 bp in size and located between nad5 and trnL2 (uaa), had a much higher A + T content (82.6%) than any other part of the mitogenome (supplementary table S2). Within the major NCR there were two highly repetitive regions (HRRs): HRR1 and HRR2, 587 and 416 bp in size, 78.% and 92.3% A + T content, respectively. HRR1 was composed of six tandem repeats, ranging from 31 to 115 bp in length (fig. 4). Repeat units 2 and 3 were identical in nucleotide composition (both 110 bp). In comparison to these two repeat units, unit 1 was 1 bp longer (111 bp) and differed in two nucleotides; unit 4 was identical in size (110 bp) and differed in one nucleotide, and unit 5 was 5 bp longer (115 bp) and differed in seven nucleotides. Repeat unit 6 was considerably truncated (31 bp), with approximately 80 bp missing at the 3′-end. HRR2 also possessed six DNA repeats, rich in A + T. Repeat units 2–5 were identical (78 bp), whereas the truncated units 1 and 6 were 64 and 40 bp long, respectively. The consensus repeat units of HRR1 (2–3) and HRR2 (2–5) were capable of forming stem-loop structures: −9.48 kcal/mol for the former and −12.24 kcal/mol for the latter (fig. 4). Stem-loop secondary structure of the repeat unit was also reported in polyopisthocotylids, and was assumed to be associated with the origin of replication (Park et al., Reference Park, Kim, Kang, Kim, Eom and Littlewood2007; Zhang et al., Reference Zhang, Wu, Xie and Li2012).

Fig. 4. Illustration of highly repetitive regions in the major non-coding region of the Dactylogyrus lamellatus mitochondrial genome. ‘HRR1’ and ‘HRR2’ represent two different repeat regions, both containing six repeat units (repeat 1–6). Sequence alignment of the HRR1 repeat units is shown below, and HRR2 is above. The length of each repeat unit is shown on the right of the alignments. Consensus sites are highlighted with exclamation point symbols and non-consensus sites with asterisks. Logos represent the information content of the aligned sequences at a position and the relative frequency of a base. Secondary structure of the consensus repeat units of HRR1 (2–3) is shown below the alignment, and HRR2 (2–5) above the alignment.
Usually, invertebrate mtDNA genomes possess one or two major NCR(s), rich in A + T content, and hence referred to as ‘AT-rich regions’ (Lunt et al., Reference Lunt, Whipple and Hyman1998). Tandem repeats (TRs), presumed to be a consequence of strand slippage during replication (Levinson & Gutman, Reference Levinson and Gutman1987), are common in the major non-coding region of mitogenomes of Cestoda (von Nickisch-Rosenegk et al., Reference von Nickisch-Rosenegk, Brown and Boore2001; Duan et al., Reference Duan, Gao, Hou, Zhang, Liu, Gao, Guo, Yue, Su, Fu and Wang2015). Zhang et al. (Reference Zhang, Wu, Xie and Li2012) have proposed a hypothesis that, within the Monogenea, monopisthocotylids can be distinguished from polyopisthocotylids by having fewer and smaller (in size) TRs in the major NCR. However, the NCR of D. lamellatus was pronouncedly longer than NCRs in other monopisthocotylid mitogenomes published to date. The structure of the NCR (large in size and containing two HRRs) and the pattern of the two HRRs (rich in iterations) are similar to those observed in a polyopisthocotylid species, Pseudochauhanea macrorchis (Zhang et al., Reference Zhang, Wu, Xie and Li2012), which indicates that we can reject that hypothesis. Due to the variability in the iteration number and polymorphisms within and between populations, TRs have been exploited extensively in population genetics studies in a broad range of animal species, including insects, fish and molluscs (Shui et al., Reference Shui, Han, Gao and Miao2008; Liu et al., Reference Liu, Kurokawa, Sekino, Tanabe and Watanabe2014; Atray et al., Reference Atray, Bentur and Nair2015). Hence, these tandem repeats may provide a novel molecular marker for further studies on systematics and population genetics of D. lamellatus.
Phylogeny and gene order
Both phylograms (BI and ML) had very high statistical support for the branch topology: with one exception (99), all bootstrap support values were 100 and all Bayesian posterior probabilities (BPP) were 1.0. Since both phylograms had identical topology, only the latter is shown (fig. 3A). The tree topology indicated the existence of two major clades: Monopisthocotylea subclass (Gyrodactylidae, Capsalidae and Dactylogyridae) and Polyopisthocotylea subclass (Microcotylidae and Chauhaneidae). Monopisthocotylea subclass was also divided into two major clades: one containing only the Gyrodactylidae family, and the other containing Capsalidae and Dactylogyridae families. This supports early taxonomic classifications: Dactylogyridae and Capsalidae were originally placed together within the order Dactylogyridea, whereas Gyrodactylidae was placed within Gyrodactylidea, and later reassigned to the corresponding superorders Dactylogyria and Gyrodactylia (Lebedev, Reference Lebedev1988). These classifications mostly relied on morphological characteristics, such as the number of edge hooks, of which there are 14 in Dactylogyridae and Capsalidae, and 16 in Gyrodactylidae. Several other characteristics specific for Gyrodactylidae further support the hypothesis that this family is evolutionarily distant from the Capsalidae and Dactylogyridae cluster: viviparity, the absence of a larval stage, the absence of eyes and the corona of chitinous hooks placed within the copulatory organ (Bychowsky, Reference Bychowsky1957).
The results of our phylogenetic analysis are in agreement with the genetic relationships inferred from previous studies that used mitogenomic data to reconstruct the phylogenetic relationships among monogeneans (Bachmann et al., Reference Bachmann, Fromm, Patella de Azambuja and Boeger2016; Zhang et al., Reference Zhang, Zou, Zhou, Wu, Li and Wang2016; Zou et al., Reference Zou, Zhang, Li, Zhou, Wu and Wang2016). Remarkably, all (but one) nodes of our phylogenetic tree had the maximum possible statistical support (BP = 100, BPP = 1.0), which was not the case in a very recent study that also used 36 concatenated genes for the analysis (Bachmann et al., Reference Bachmann, Fromm, Patella de Azambuja and Boeger2016). This might be a consequence of the selection of outgroups: whereas Bachmann et al. (Reference Bachmann, Fromm, Patella de Azambuja and Boeger2016) used two Cestoda species, we used two Tricladida (Turbellaria) species. To test this hypothesis, we have replaced our two outgoup sequences with these two Cestoda sequences and conducted another phylogenetic test on our dataset: the topology was identical, but statistical support values were somewhat lower (supplementary fig. S3). In order to infer the correct topology, an outgroup should be a taxonomic group that diverged before the last common ancestor of the ingroup (Pearson et al., Reference Pearson, Hornstra, Sahl, Schaack, Schupp, Beckstrom-Sternberg, O'Neill, Priestley, Champion and Beckstrom-Sternberg2013). As Cestoda appears to be younger, and Turbellaria older, than Monogenea (Park et al., Reference Park, Kim, Kang, Kim, Eom and Littlewood2007; Perkins et al., Reference Perkins, Donnellan, Bertozzi and Whittington2010), rooting with Tricladida (Turbellaria) species should result in a topology more closely resembling the actual evolutionary history of these taxonomic groups. Most importantly, as none of these studies had a Dactylogyrinae mitogenome sequence at their disposal, our results markedly improve the resolution of monogenean relationships.
Although several researchers suggested that Ancyrocephalinae should be elevated to the family level (Bychowsky & Nagibina, Reference Bychowsky and Nagibina1978; Lebedev, Reference Lebedev1988), our phylogenetic results indicate a sister-group relationship between Ancyrocephalinae and Dactylogyrinae with maximum support value, which adds support to the revision of Kritsky & Boeger (Reference Kritsky and Boeger1989). Beyond these, mtDNA gene order and transformational pathway analysis in this study has also provided additional supporting evidence for the close relationship between these two subfamilies. Pairwise comparisons of the mitochondrial gene order among seven taxa revealed that, based on the dissimilarity value (breakpoint algorithm), D. lamellatus was the most similar to T. nebulosi. As expected, D. lamellatus was the most dissimilar from the three Polyopisthocotylea species: Microcotyle sebastis (Goto, 1894), P. halichoeres and P. macrorchis (table 2), mostly due to the pronounced differences in gene order at the cox3–nad5 junction between monopisthocotylids and polyopisthocotylids (Zhang et al., Reference Zhang, Wu, Li, Zhao, Xie and Li2014b) (also see fig. 3B). The transformational pathway from D. lamellatus to its sister taxon T. nebulosi requires one transposition event; a single transposition and two coupled transposition events are required to Neobenedenia melleni (MacCallum, 1927); and two coupled long-distance transpositions are required to Paragyrodactylus variegatus (You et al. Reference Ye, King, Cone and You2014) (supplementary fig. S4). trnL2 (uaa) abutted upstream of the trnG (ucc) in the mitogenome of D. lamellatus, but in other monopisthocotylids it is usually located between trnS2 (uga) and trnR (ucg). Aglaiogyrodactylus forficulatus genome, where trnL2 (uaa) is located between trnR (ucg) and nad5, is another exception. Apart from the position of trnL2 (uaa), the gene order of D. lamellatus was identical to that of T. nebulosi. From N. melleni, it differed with respect to the position of NCR and trnQ (uug) (fig. 3B).
Table 2. Pairwise breakpoint comparison of mitochondrial DNA gene orders among seven monogenean species, based on the order of all 37 elements (36 genes plus NCR). Taxa with multiple NCRs were not used for the analysis. Scores indicate the dissimilarity between gene orders, where ‘0’ represents an identical gene order. N is the number identifying the taxon in the pairwise comparisons.

In conclusion, we have sequenced and analysed the complete mitogenome of D. lamellatus, which contains the standard 22 tRNA genes, 2 rRNA genes and 12 protein-encoding genes (PCGs). There are two highly repetitive regions in the NCR. Using concatenated nucleotide sequences of all 36 genes (12 PCG, 2 rRNA and 22 tRNA genes), the phylogenetic analysis performed using Bayesian inference and maximum likelihood methods revealed that the two dactylogyrids, D. lamellatus (Dactylogyrinae) and T. nebulosi (Ancyrocephalinae), are very closely related to each other. These two then form a sister group with Capsalidae, and finally, this cluster further forms a sister group with Gyrodactylidae. This result is in agreement with some early traditional classifications and is further supported by morphological characteristics. The phylogenetic affinity between Dactylogyrinae and Ancyrocephalinae is further confirmed by the similarity in their mitochondrial gene arrangement. As many lineages of the class Monogenea are still underrepresented (such as Dactylogyridae and Chauhaneidae), or not represented at all (such as Diplectanidae, Monocotylidae and Diplozoidae), our results do not provide a comprehensive phylogenetic hypothesis for the Monogenea. In order to resolve the evolutionary relationships among the monogeneans with confidence, a much larger number of mitogenomic sequences will have to be available. Hence, the publication of this mitogenome will lend support to future molecular, evolutionary and population studies of D. lamellatus and related monogenean parasites.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S0022149X17000578
Acknowledgements
The authors would like to thank Dr Bao J. Yang for kindly providing the Dactylogyrus lamellatus sample. We would also like to thank the editor and the two anonymous reviewers for the time they have invested in reviewing our manuscript.
Financial support
This work was funded by the Earmarked Fund for China Agriculture Research System (CARS-46-08); the National Natural Science Foundation of China (31172409, 31272695); and the Major Scientific and Technological Innovation Project of Hubei Province (2015ABA045).
Conflict of interest
None.
Ethical standards
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional guides on the care and use of laboratory animals.







