Introduction
Despite an increasing reliance of biomedical research on high-throughput screening methods, the application of quantitative imaging techniques to analyze protein behavior in the context of living cells remains vitally important. These live-cell imaging approaches have greatly benefitted from recent improvements in the photophysical qualities of the genetically encoded fluorescent proteins (FPs) [Reference Day and Davidson1]. The favorable spectral characteristics of the optimized FPs enable their use in Förster resonance energy transfer (FRET) microscopy to achieve the nanometer-scale resolution necessary to detect protein interactions in living cells. FRET microscopy measures the direct transfer of excitation energy from one fluorophore (the donor) to other nearby molecules (acceptors). Because FRET occurs through near-field electromagnetic dipole interactions, the efficiency of energy transfer decreases as the inverse of the sixth power of the distance separating the fluorophores [Reference Periasamy and Day2]. This limits FRET to separation distances of less than about 8 nm, making it an ideal tool to investigate cellular biochemical networks.
There are many different FRET microscopy approaches, broadly divided into methods that detect the sensi-tized emission from the acceptor and methods that detect the effect of energy transfer on the donor fluorophore [Reference Periasamy and Day2]. A key requirement for efficient energy transfer is a strong overlap between the donor emission spectrum and the absorption spectrum of the acceptor (Figure 1). A consequence of this overlap, however, is spectral bleedthrough (SBT) background that originates both from the direct excitation of the acceptor at the donor excitation wavelengths (arrow, Figure 1) and from the donor emission signal that bleeds into the FRET detection channel (hatching, Figure 1). Therefore, the quantification of FRET by measurement of sensitized emission from the acceptor requires correction methods to remove these different SBT background components [Reference Periasamy and Day2]. In contrast, the signal in the donor channel is not contaminated by SBT (Figure 1). Thus, methods that directly measure the effect of energy transfer on the donor signal do not require corrections and can be the most accurate way to quantify FRET. In this article, the use of fluorescence lifetime imaging microscopy (FLIM) to quantify the change in donor lifetime that occurs with FRET is demonstrated, and the advantages and the limitations of the approach are discussed.

Figure 1 The SBT between donor and acceptor fluorohores. The excitation and emission spectra for cyan (donor) and yellow FPs (acceptor) are shown. The dashed boxes indicate typical setup for the detection of donor and FRET (acceptor) signals. The arrow indicates the direct acceptor excitation at the donor excitation wavelength, and the hatching shows the donor SBT into the FRET channel.
Fluorescence Lifetime Imaging Microscopy
The fluorescence lifetime is the average time that a popula-tion of fluorophores spends in the excited state before returning to the ground state, an event usually accompanied by the emission of photons. The fluorescence lifetime is an intrinsic property of a fluorophore, and changes in the lifetime can provide information about the microenvironment surrounding the fluorophores. For example, energy transfer is a quenching pathway that depletes excited-state energy from the donors, decreasing the average lifetime. This change in the fluorescence lifetime can be accurately quantified by FLIM. This approach is particularly useful for biological applications because measurements made in the time-domain are not affected by variations in probe concentration, by changes in excitation intensity, nor by light scatter in the sample, all of which can impact steady-state intensity measurements.
The FLIM techniques are broadly subdivided into time domain and frequency domain (FD) methods. The physics that underlies these two methods is identical, and they differ only by the analysis of the measurements [Reference Periasamy and Clegg3]. The frequency domain (FD) FLIM approach described here uses a laser modulated at high frequencies (10–140 MHz) to excite the fluorophores. Because of the lifetime of the excited state, there is a phase delay (Φ) and a change in the modulation (M) of the emission signal relative to the excitation waveform (Figure 2). Fluorescence lifetimes are directly determined from both the Φ and the M of the emission signal simultaneously measured at many different modulation frequencies (ω). The data are then represented using phasor plots that do not require fitting algo-rithms or any a priori knowledge of the system.
Figure 2 The FD FLIM system. The excitation source for the FD FLIM system in this study is a 440 nm diode laser that is modulated by the ISS FastFLIM module at the fundamental frequency of 10 MHz and 13 harmonic frequencies (ω). The modulated laser is coupled to the ISS scanning system that is attached to an Olympus IX71 microscope with an environmentally controlled stage. The emission signals from the specimen travel through the scanning system (de-scanned detection), and are routed by a beam splitter through the donor and acceptor emission channel filters to two identical APDs.
Materials and Methods
DNA preparation, cell culture, and transfection
The cDNAs encoding the monomeric (m) Turquoise, mCerulean3, and mVenus FPs, were obtained from Michael Davidson (Florida State University, Tallahassee, FL). Standard recombinant DNA methods were used to generate the plasmids encoding the FRET standard proteins and fusion proteins described below. All plasmid inserts were confirmed by direct seq-uencing. The plasmid DNAs were used in transfection of mouse pituitary GHFT1 cells by electroporation as described previously [Reference Day4]. Immediately after elec-troporation, the cells are recovered and diluted in phenol red-free tissue culture medium containing serum. The cells are transferred into sterile two-well-chambered coverglasses (Lab-Tek II, Thermo Scientific), which are maintained at 37ºC and 5% CO2 prior to the FLIM analysis.
FD FLIM measurements
Lifetime measurements are made using the ISS Alba FastFLIM system (ISS Inc., Champaign, IL) coupled to an Olympus IX71 microscope (Figure 2). The microscope is equipped with a 60 × 1.2 numerical aperture water-immersion objective lens, an objective warmer, and a stage-top environmental control system (Pathology Devices, Inc.) to maintain the temperature and CO2 levels of the sample. For FRET studies using the cyan and yellow FPs, the 5 mW 440 nm diode laser was modulated by the Alba FastFLIM system at a fundamental frequency of 10 MHz, with additional measurements at 13 harmonics (10–140 MHz). The modulated laser is coupled to the confocal scanning system that is controlled by the VistaVision software (Build 218, ISS Inc., Champaign, IL). The fluorescence signals emitted from the specimen are routed by a 495 nm long-pass beam splitter through the 530/43 nm (acceptor emission) and 480/40 (donor emission) band-pass emission filters, and the signals are detected using two identical avalanche photodiodes (APD).
Calibration and data acquisition
The system is calibrated with 50 μM Coumarin 6 dissolved in ethanol, which has a lifetime of 2.5 ns and is used as a reference for the software. The calibration is verified by measurement of the lifetime of 2 mM HPTS (8-hydroxypyrene-1,3,6-trisulfonic acid) dissolved in phosphate buffer at pH 7.8 (reference lifetime of 5.3 ns). For live-cell FD FLIM measurements from cells producing the FP-labeled proteins, the cells are first identified by epi-fluorescence microscopy. Frequency domain FLIM measurements are then acquired from the selected cells. The field of view used here is 256×256 pixels, and frame averaging was used to accumulate about 200 peak counts per pixel, which typically requires about 45 seconds. The FLIM images were analyzed with the VistaVision software by selecting regions of interest (ROI, typically 10 μm2) with average intensity >100 counts.
Results
Verification of the calibration
Following calibration, FLIM measurements of Coumarin 6 were obtained from a 256 × 256 pixel field of view (65,536 pixels). The Φ and M of the emission signal at each image pixel was acquired simultaneously at the fundamental frequency (10 MHz) and 13 harmonics (20–140 MHz). Then lifetime measurements were acquired from a second reference standard, 10 mM HPTS. The multi-frequency response curves in Figure 3 show both the Φ and M of the emission signal for Coumarin 6 and HPTS at each frequency, and the chi-square values determine the quality of the data fit.
Figure 3 Multi-frequency response curves for Coumarin 6 and HPTS. The phase delays (Φ) and modulation ratios (M) of the emission signals for Coumarin 6 and HPTS were measured at the fundamental frequency (10 MHz) and 13 harmonics (ω=10–140 MHz). The average lifetime (τ) and chi-square values for fitting the data are shown.
The fluorescence lifetime of both Coumarin 6 and HPTS were then analyzed using the phasor plot method [Reference Jameson5–Reference Hinde7]. The phasor plot is a simple geometric representation of the Φ and M of the emission signal. The time delays from each image pixel are transformed into vectors with the length determined by M and the angle determined by the Φ (Figure 4). The lifetime distribution for all image pixels at a particular frequency is then plotted relative to a universal semicircle, with the incidence of lifetime values indicated from blue (highest) to red to yellow (lowest; see Figure 4). The semicircle is universal in the sense that the lifetime distribution for any species with a single-exponential decay will fall directly on the semicircle irrespective of the lifetime or modulation frequency [Reference Redford and Clegg6]. Here, the lifetime distribution for Coumarin 6 falls directly on the universal semicircle at a position corresponding to an average lifetime of 2.5 ns (Figure 4). In contrast, the lifetime distribution for HPTS is shifted to the left along the semicircle relative to Coumarin 6, reflecting its longer lifetime. Again, the distribution falls directly on the semicircle, indicating a single lifetime component of 5.3 ns (Figure 4).
Figure 4 A composite phasor plot showing the distributions of fluorescence lifetime for Coumarin 6 and HPTS. For Coumarin 6, the centroid of the distribution falls on the universal semicircle, indicating a single exponential lifetime of 2.5 ns. For HPTS, the distribution for the longer lifetime probe is shifted to the left along the semicircle. The centroid of the distribution falls on the semicircle, indicating a single exponential lifetime of 5.3 ns.
Optimized donor FPs for FRET-FLIM
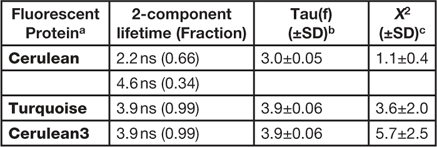
Here, the FLIM imaging method is demonstrated in living cells expressing the "FRET standard" proteins. The use of the FRET standards provides a straightforward approach to verify both the biological model and the imaging system used for the measurement of FRET. The original FRET standard fusion proteins consist of monomeric (m)Cerulean directly coupled to mVenus through linkers of different lengths [Reference Koushik8]. However, because mCerulean has more than one fluorescence lifetime (Table 1), it was replaced with the improved variants of mCerulean, mCerulean3 [Reference Markwardt9], or mTurquoise [Reference Goedhart10]. These newer variants have reduced photoswitching behavior and improved photostability compared to mCerulean. Most important for FLIM measurements, both of the improved variants have a single component lifetime (Table 1), which simplifies the lifetime analysis for FRET experiments described below. We find that mTurquoise and mCerulean3 perform equally well for FRET-FLIM studies [Reference Siegel11].
Table 1 FD FLIM analysis of the variants of Cerulean.
a Expressed in cells at 37º, n = 10 or more.
b Tau(f) is the average lifetime.
c Chi-square for the fit of measurements at 12 frequencies from 10 to 120 MHz.
FLIM measurements of the FRET standards
The fusion protein consisting of mTurquoise coupled directly to mVenus through the five-amino acid (5aa) linker serves as a high FRET efficiency standard. Because the fluorescence lifetime can be very sensitive to the local environment of the donor fluorophore, a similar fusion protein consisting of mTurquoise linked to a mutated mVenus, called Amber, was also made [Reference Koushik8]. The mutation converts the chromophore tyrosine to a cysteine producing a non-fluorescent form of Venus that folds correctly, but does not act as a FRET acceptor, providing an accurate measurement of the unquenched donor lifetime. Finally, a fusion protein consisting of mTurquoise linked to mVenus through the 229-amino acid tumor necrosis factor receptor associated factor (TRAF) domain (Turquoise-TRAF-Venus) provides a low FRET efficiency standard [Reference Thaler12].
The FRET efficiency (E FRET) is determined from the ratio of the unquenched donor lifetime (τD; Turquoise-5aa-Amber) to the quenched donor lifetime in the presence of the acceptor (τDA; Turquoise-5aa-Venus, Turquoise-TRAF-Venus):
 $$E_{{FRET}} =1{\minus}{{\tau _{{DA}} } \over {\tau _{D} }}$$
$$E_{{FRET}} =1{\minus}{{\tau _{{DA}} } \over {\tau _{D} }}$$
The composite phasor plot in Figure 5 compares the unquenched and quenched donor lifetimes for the different FRET standard fusion proteins produced in living cells. The intensity images obtained from representative cells expressing the different FRET standard fusion proteins are shown in the top panels of Figure 5, and the average lifetime measurements for each FRET standard are presented in Table 2. The average unquenched donor lifetime for Turquoise-5aa-Amber expressed in cells was 3.9 ns (Table 2), and the phasor plot shows it is best fit to a single-component decay (Figure 5A). The lifetime distribution for the Turquoise-TRAF-Venus fusion protein can be distinguished from that of the unquenched donor (Figure 5B) and is shifted to the right along the semicircle indicating a shorter lifetime. The average donor lifetime for Turquoise-TRAF-Venus expressed in cells was 3.5 ns, which corresponds to an FRET efficiency of 10%, determined using Eq. 1 (Table 2). In sharp contrast, the lifetime distribution for the cell expressing Turquoise-5aa-Venus shows the quenched donor lifetime for the high FRET standard (Figure 5C). The lifetime distribution for the high FRET standard is shifted far to the right relative to the unquenched donor and falls inside the semicircle, indicating a multi-component lifetime. When analyzing the multi-exponential lifetimes that result from energy transfer, the E FRET is determined using the amplitude-weighted lifetime [Reference Lakowicz13]. The average amplitude-weighted lifetime for Turquoise-5aa-Venus was 2.2 ns, corresponding to an E FRET of 43% (Table 2).
Figure 5 The phasor plot analysis of the lifetimes for the FRET standards produced in living cells. (A) The intensity image and phasor lifetime distribution from a cell expressing Turquoise-5aa-Amber (unquenched donor; the calibration bars are 10 μm), indicating a single exponential lifetime of 3.9 ns (Table 2). (B) The intensity image and phasor lifetime distribution from a cell expressing Turquoise-TRAF-Venus, indicating an average lifetime of 3.6 ns (Table 2). (C) The intensity image and phasor lifetime distribution from a cell expressing Turquoise-5aa-Venus demonstrating a multi-component lifetime. The average amplitude-weighted average lifetime was 2.2 ns (Table 2).
Table 2 FD FLIM analysis of the FRET standards.
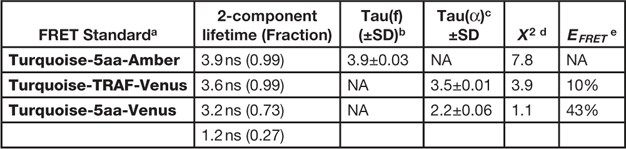
a Expressed in cells at 37º, n=6 cells.
b Tau(f) is the average lifetime.
c Tau(α) is the amplitude-weighted lifetime.
d Chi-square for the fit of measurements at 12 frequencies from 10 to 120 MHz.
e Determined using Eq. 1
FLIM measurements of protein-protein interactions
Because FLIM directly measures the quenching of the donor resulting from FRET, and does not require correction for SBT or assumptions in the data analysis, the interpretation of intermolecular FRET measurements of protein-protein interactions is simplified compared to other methods [Reference Periasamy and Day2–Reference Hinde7]. However, because the donor- and acceptor-labeled proteins are produced independently of one another in intermolecular FRET experiments, there will be a different ratio of the labeled proteins in each co-transfected cell. The donor-to-acceptor ratio influences the E FRET because cells expressing predominately donor-labeled proteins will have little or no FRET, whereas cells with an excess of acceptor have the potential for high FRET signals [Reference Day4,Reference Siegel11]. Therefore, the ratio of donor- to acceptor-labeled proteins must be accounted for in the analysis of data from intermolecular FRET experiments. The FLIM system used here has two-channels that simultaneously detect the emission signals from the donor and acceptor (Figure 2). By measuring the mean intensity in both the donor (I D) and the acceptor (I A) channels, the ratio of the acceptor- to the donor-labeled proteins can be qualitatively determined. Then, the measurement of the quenched donor lifetime in the same ROI allows the determination of E FRET (Eq. 1) at that I A/I D ratio.
Here the FRET-FLIM approach is used to quantify the heterologous protein interactions between the basic-leucine zipper (BZip) domain of the transcription factor C/EBPα and the heterochromatin protein 1 alpha (HP1α). In earlier studies, we demonstrated that HP1α and C/EBPα interact, but only when the proteins are bound to heterochromatin [Reference Siegel11]. For these intermolecular FRET measurements, the lifetime for the BZip domain labeled with Turquoise (the unquenched donor) was determined in regions of heterochromatin (typically 5 to 10 ROI per cell nucleus) for 10 different cells, yielding an average unquenched donor lifetime of 3.89±0.08 ns. Intensity images and lifetime measurements were then acquired from cells co-producing the mTurquoise-BZip domain and HP1α proteins labeled the acceptor FP, Venus (Figure 6A). As a control for potential non-specific interactions, measurements were also made from cells producing the mTurquoiseN1 protein (localized in both cytoplasm and nucleus) and Venus-HP1α (nuclear, see Figure 6B). The lifetime map in Figure 6B demonstrates the quenched lifetimes of the Turquoise-BZip domain co-expressed with the Venus-labeled HP1α, indicated by cooler colors on the look-up table. In contrast, the lifetime of the TurquoiseN1 protein was not changed when co-expressed with Venus-HP1α (Figure 6B). To characterize the interaction between mTurquoise-BZip domain proteins and Venus-HP1α, the E FRET was determined from lifetime measurements of 15 cells co-producing the proteins at different I A/I D ratios (Figure 6C). The results show the dependence of E FRET between BZip domain and HP1α in regions of heterochromatin on the I A/I D ratio, indicating the close proximity of fluorophores labeling the proteins (Figure 6C). In contrast, there was no change in the lifetime of mTurquoiseN1 with increasing Venus-HP1α.
Figure 6 FRET-FLIM analysis of the heterologous interactions between the C/EBPα BZip domain and HP1α. (A) The intensity images for the nucleus of a cell expressing both the mTurquoise-BZip domain and mVenus-HP1α acquired in the acceptor channel, donor channel (the calibration bars are 10 μm), and the corresponding lifetime map with I A/I D ratio indicated. (B) The intensity images for a cell expressing both the mTurquoiseN1 (localized throughout the cytoplasm and nucleus) and mVenus-HP1α (nuclear) acquired in the acceptor channel, donor channel, and the corresponding lifetime map with the I A/I D ratio indicated. (C) FLIM was used to measure the donor lifetime in multiple ROI for each cell, and the FRET efficiency (%) was determined. Each point represents the average E FRET (±SD) at the average I A/I D (±SD) for multiple ROI in individual cells expressing the indicated donor- and acceptor-labeled proteins. Reprinted from [Reference Day4] with permission from Elsevier.
Discussion
The development of quantitative imaging techniques to visualize cellular events inside living cells continues to provide critical tools for biomedical research. The combination of FD FLIM measurements and phasor analysis described here enables a variety of quantitative approaches to interrogate events inside living cells. For example, in addition to the intermolecular FRET measurements illustrated above, the FLIM approach with phasor analysis is an outstanding method to monitor the activities of FRET-based biosensor probes that report intracellular signaling events [Reference Hinde7]. A distinct advantage of the phasor plot approach is that multi-component lifetimes that occur in populations of proteins involved in FRET are immediately evident from lifetime distributions that fall inside the universal semicircle. As can be seen in Figure 5C, this is also true for the linked FRET standard proteins, as well as similarly designed biosensor probes. The reason the linked probes also have multi-component lifetimes is that the FPs rotate slowly relative to their fluorescence lifetime, so there is negligible change in their relative orientations during the excited-state lifetime. This will give rise to heterogeneity in population of dipole orientations, leading to a distribution of E FRET for the linked FRET standard [Reference Vogel14].
A general limitation of the FRET-FLIM approach described here is the necessity of producing the exogenous proteins of interest labeled with the FPs inside living cells, which can potentially cause artifacts associated with overexpression. This concern is not unique to FLIM; it is shared with many different types of cellular assays and requires careful control experiments. For example, where possible it is important to verify that FP-labeled proteins co-localize with their endogenous counterparts using immunostaining. It is also critical to verify that the FP-labeled proteins or biosensor probes accurately report cellular events using approaches such as mutagenesis. The overexpression of the FP-labeled proteins or biosensor probes can potentially interfere with the cellular processes that they are designed to measure. The FPs described here have been engineered to optimize their characteristics for expression and imaging in living cells and are well tolerated in living systems [Reference Day and Davidson1]. In addition, the imaging system has single-molecule detection sensitivity, which has allowed us to complement the FRET-FLIM with FCCS measurements [Reference Siegel11]. Significantly, the FLIM approach is not limited to exogenously labeled proteins. The measurement of intrinsic autofluorescence signals from living cells by FLIM and the analysis by phasor plot have been used to generate metabolic fingerprints from cells, allowing the identification of differentiating cells or cancer cells according to their metabolic state [Reference Stringari15, Reference Wright16].
Conclusion
The FD FLIM method provides a direct measurement of FRET that requires no assumptions or corrections and is among the most accurate methods for monitoring protein interactions or biosensor probe activities. This non-invasive imaging technique provides a method to verify the results obtained by high-throughput screening methods. Most important, the measurements obtained from proteins in their natural environment inside living cells provide the most physiologically relevant information about protein behavior currently available.
Acknowledgements
This research was supported in part by NIH P30DK079312 and the Indiana University School of Medicine. The author thanks Dr. Yuansheng Sun and Dr. Jeff Liao (ISS Inc., Champaign, IL) for their advice and technical support and Michael Davidson for the plasmids encoding the FPs.