


THE DIVERSITY OF THE NEMATODA
Nematoda is an ancient and biologically diverse phylum of moulting animals. They range in size from 0·2 mm to over 6 m, and can be found in most habitats, including within and on host animals and plants (Blaxter and Denver, Reference Blaxter and Denver2012). In many marine and terrestrial sediments they are the most abundant group in terms of individuals (Platonova and Gal'tsova, Reference Platonova and Gal'tsova1976), and while only approximately 23 000 species have been described (J. Hallan, unpublished; https://insects.tamu.edu/research/collection/hallan/), the true species-level diversity may be 1 million or more (Lambshead, Reference Lambshead1993). Most terrestrial plants and larger animals are associated with at least one species of parasitic nematode, and most of the human population experiences nematode parasitism during their lives (with perhaps one quarter to one third of the global population infected at any time). Estimates of the number of species of parasitic nematode per host suggest that there may be of the order of 25 000 nematode parasites just of vertebrates, most of which remain undescribed (Dobson et al. Reference Dobson, Lafferty, Kuris, Hechinger and Jetz2008). Nematodes are thus important regulators of plant and animal production. Understanding the evolutionary origins of plant and animal parasitism, and the mechanisms by which parasites locate and invade their hosts, avoid host immunity, and acquire nutrition, are important goals for not only basic, but also for medical and veterinary science. In this paper we discuss the changes in our understanding of the diversity and relationships of nematodes, and of the biology of their parasitic habits, that have been brought about by study of their genes and, increasingly, genomes.
Nematoda are part of Ecdysozoa, a superphylum of animals first defined through analyses of molecular markers (Aguinaldo et al. Reference Aguinaldo, Turbeville, Linford, Rivera, Garey, Raff and Lake1997). Support for Ecdysozoa as distinct from other groupings of protostome taxa is less strong from analyses of morphological characters (Nielsen, Reference Nielsen2001). Ecdysozoan phyla are characterized by the presence of a cuticle that is periodically moulted during the life cycle, though the specifics of the molecular nature of the cuticle and the orchestration of ecdysis differ between phyla. Other shared features adduced as evidence of relatedness between these phyla include an absence of cilia in adults, and in many members the presence of a triradiate pharynx. The Ecdysozoa in turn comprises two groups, the Panarthropoda (phyla Tardigrada, Onychophora and Arthropoda) and Cycloneuralia (Nematoda, Nematomorpha, Priapulida, Kinorhyncha and Loricifera). Within Cycloneuralia, which may be paraphyletic with respect to Panarthropoda, Nematoda are consistently placed as sisters to Nematomorpha in morphological and molecular analyses (Schmidt-Rhaesa, Reference Schmidt-Rhaesa1997; Dunn et al. Reference Dunn, Hejnol, Matus, Pang, Browne, Smith, Seaver, Rouse, Obst, Edgecombe, Sorensen, Haddock, Schmidt-Rhaesa, Okusu, Kristensen, Wheeler, Martindale and Giribet2008).
Nematomorpha are a fascinating group of obligate parasites of terrestrial (Gordioidea) and marine (Nectonematoidea) arthropods. These ‘horsehair worms’ have a parasitoid life cycle, with the larval stages residing within the body cavities of their arthropod hosts, which they kill when they emerge. The adult sexual stages are free-living in pelagic (Nectonematoidea) or sediment (Gordioidea) habitats. Infection of the next host is by ingestion of eggs, often glued to vegetation eaten by the hosts (Hanelt and Janovy, Reference Hanelt and Janovy1999). The generalized life cycle of nematomorphs is very similar to that of mermithid nematodes, which also have marine and terrestrial members, and which also infect their hosts as larvae but have free-living adult stages. The placement of a phylum wherein all members are parasites as sister to all of Nematoda raises the interesting question of whether the ancestor to all nematodes was a parasite (with biology similar to nematomorphs or mermithids), and that the extant free-living groups in Nematoda are reversions from this ancestrally parasitic state. This question has historically been answered in the negative, and molecular data support this conclusion, because free-living nematodes arise basally to Mermithida in Nematoda. The similarity in lifestyle is thus most likely to be homoplasious (i.e. has arisen independently by convergent evolution).
The molecular systematics of the Nematoda have been explored for nearly 20 years (Blaxter et al. Reference Blaxter, De Ley, Garey, Liu, Scheldeman, Vierstraete, Vanfleteren, Mackey, Dorris, Frisse, Vida and Thomas1998; Kampfer et al. Reference Kampfer, Sturmbauer and Ott1998), and comprehensive analyses of the breadth of diversity of the phylum now converge on a stable phylogeny (Meldal et al. Reference Meldal, Debenham, De Ley, De Ley, Vanfleteren, Vierstraete, Bert, Borgonie, Moens, Tyler, Austen, Blaxter, Rogers and Lambshead2007; Holterman et al. Reference Holterman, Karssen, van den Elsen, van Megen, Bakker and Helder2009; van Megen et al. Reference van Megen, van den Elsen, Holterman, Karssen, Mooyman, Bongers, Holovachov, Bakker and Helder2009; Bik et al. Reference Bik, Lambshead, Thomas and Lunt2010). These analyses have largely used the nuclear small subunit ribosomal RNA gene (nSSU), as it combines features of conservation and change that are informative over deep timescales. New genomic data are being brought to bear on the phylogenetics of Nematoda, and revisions of the tree may still be necessary (see below). While most traditional analyses suggested a bipartite division of the phylum, into ‘Adenophorea’ (largely marine, but also including terrestrial plant and animal parasites) and ‘Secernentea’ (largely terrestrial, and including many animal and plant parasites), the molecular analyses show three major divisions (Fig. 1A). The ‘Adenophorea’ are split between these three divisions, and ‘Secernentea’ are a subgroup of one. The new phylogeny was sytematized by De Ley and Blaxter (Reference De Ley, Blaxter and Lee2002, Reference De Ley, Blaxter, Cook and Hunt2004).

Fig. 1. The phylogenetic structure of the Nematoda and the origins of parasitism (A) A cartoon of the phylogenetic structure of the Nematoda, based on nuclear small subunit ribosomal RNA analyses and interpretation of taxon relationships derived from morphology (De Ley and Blaxter, Reference De Ley, Blaxter, Cook and Hunt2004; Blaxter and Denver, Reference Blaxter and Denver2012). Taxon systematic names are given for the major nodes in the phylogeny. Clades I, II, C, III, IV and V were first defined in Blaxter et al. (Reference Blaxter, De Ley, Garey, Liu, Scheldeman, Vierstraete, Vanfleteren, Mackey, Dorris, Frisse, Vida and Thomas1998). Helder and colleagues revised the numbering of clades (Holterman et al. Reference Holterman, van der Wurff, van den Elsen, van Megen, Bongers, Holovachov, Bakker and Helder2006; van Megen et al. Reference van Megen, van den Elsen, Holterman, Karssen, Mooyman, Bongers, Holovachov, Bakker and Helder2009), and their schema is given in smaller Arabic numerals beneath the relevant branches. For each ordinal/subordinal group named, the ecosystem and trophic habits are indicated by small icons. For the major clades, the numbers of published genomes, genomes in progress and the proportion of named species (Hallan, Reference Hallan2007) are given. (B) The utility of large scale nematode genome data for phylogenetic analyses. A phylogeny of Nematoda derived from 181 protein coding genes from 23 nematode species, and four ecdysozoan taxa as outgroup. The alignment was subjected to analysis with PhyloBayes (Lartillot et al. Reference Lartillot, Lepage and Blanquart2009), and all nodes had posterior probability of 1·00. The major clades in Rhabditida are resolved, and Enoplia is recovered at the base of Nematoda. The figure is adapted from Blaxter et al. (Reference Blaxter, Koutsovoulos, Jones, Kumar, Elsworth, Cotton, Hughes and Olson2014).
Nematoda comprises the subclasses Enoplia, Dorylaimia and Chromadoria (De Ley and Blaxter, Reference De Ley, Blaxter and Lee2002, Reference De Ley, Blaxter and Lee2004). In nSSU analyses the branching order of these three groups is unresolved, though there are hints that Enoplia may be the earliest-branching of the three (van Megen et al. Reference van Megen, van den Elsen, Holterman, Karssen, Mooyman, Bongers, Holovachov, Bakker and Helder2009; Blaxter et al. Reference Blaxter, Koutsovoulos, Jones, Kumar, Elsworth, Cotton, Hughes and Olson2014). The inability of nSSU to robustly distinguish the branching order and thus the root of the phylum is due to lack of strong signal, exacerbated by the phylogenetic distance to the nearest outgroup taxa (other Ecdysozoa, which likely last shared a common ancestor well before the Cambrian, over 540 My ago).
It is generally argued that Nematoda has a marine origin (see Fig. 1A). The Enoplia are largely marine, and mostly free-living. They are the commonest nematodes in marine sediments, and dominate deep-sea ecosystems where they feed on diatoms and marine algae. Members of Enoplia are also found in brackish and fresh water, and on land, including plant parasites. The Dorylaimia are freshwater or terrestrial nematodes and include major groups of plant and animal parasites. The Chromadoria includes a large number of marine groups, and a major terrestrial radiation that includes plant and animal parasites. In Chromadoria the terrestrial taxa appear to have arisen from marine ancestors, but the situation in the other subclasses is less clear. Dorylaimia has few truly marine taxa, and in Enoplia molecular phylogenies place the terrestrial/freshwater Triplonchida as sister to the remaining (marine) Enoplida.
THE MULTIPLE ORIGINS OF PARASITISM WITHIN THE NEMATODA
Nematodes exhibit a wide range of relationships with other species. Parasitism is a common way of life, and a large proportion of nematode species may be parasites. Poulin has usefully classified the different kinds of parasitic relationships between species into a spectrum of life-habit modes (Poulin, Reference Poulin2011; Poulin and Randhawa, Reference Poulin and Randhawa2013). Some relationships are phoretic: the nematodes use another species to aid dispersal to new sites (Bovien, Reference Bovien1937). Phoretic associations can be very specific, as in Rhabditis stammeri, associates of burying beetles (Nicrophorus spp.), which have a complex response to the beetle life cycle that assures their presence in emerging adults by entering and diapausing in the hind guts of mature larvae before they pupate (Richter, Reference Richter1993). Other phoretic relationships are less specific, and dispersal stages can be found attached to many different transport hosts. In most cases, the dispersal stage is a third stage juvenile (J3, or L3 for larva). The costs to the phoretic host are hard to measure, but may be significant in heavily colonized hosts, or where the associate extracts some nutrition from its carrier. Many plant parasitic nematodes, particularly the migratory endoparasites, could be classified as microherbivores (Poulin and Randhawa, Reference Poulin and Randhawa2013), as their ecology is similar to that of an ungulate browsing on bushy plants. However these nematodes do induce specific cell responses in host plants, and thus the relationship is more than just browsing. Some intestinal parasites, such as the Rhigonematida of millipedes (Hunt, Reference Hunt1996), feed on gut contents or other nematode parasites rather than on host tissue, and might even be classed as commensals.
Independent origins of the parasitic habit can be validated by molecular phylogenetic placement of parasitic taxa and their free-living relatives (Blaxter et al. Reference Blaxter, De Ley, Garey, Liu, Scheldeman, Vierstraete, Vanfleteren, Mackey, Dorris, Frisse, Vida and Thomas1998; Dorris et al. Reference Dorris, De Ley and Blaxter1999). With the current available molecular data we can define three origins of plant parasitism, 10 of parasitism of a wide range of non-vertebrates and five of parasitism of vertebrate hosts across the three subclasses (Fig. 1 and Table 1). Many additional events of acquisition of parasitic lifestyles could be proposed. For example, Sudhaus has suggested at least 20 independent events of acquisition of parasitism of insects in Nematoda (Sudhaus, Reference Sudhaus2008). Enoplia has the fewest parasitic species, while Chromadoria (and Rhabditida within Chromadoria) has the most. All parasitic groups appear to have a terrestrial or limnic origin. The sole enoplian animal parasite, Ironus macrocephalum (Ironidae), was described from the earthworm Pheretima wendessiana in New Guinea (Pierantoni, Reference Pierantoni1916). Other Ironidae are freshwater species.
Table 1. Origins of parasitism in the Nematoda a
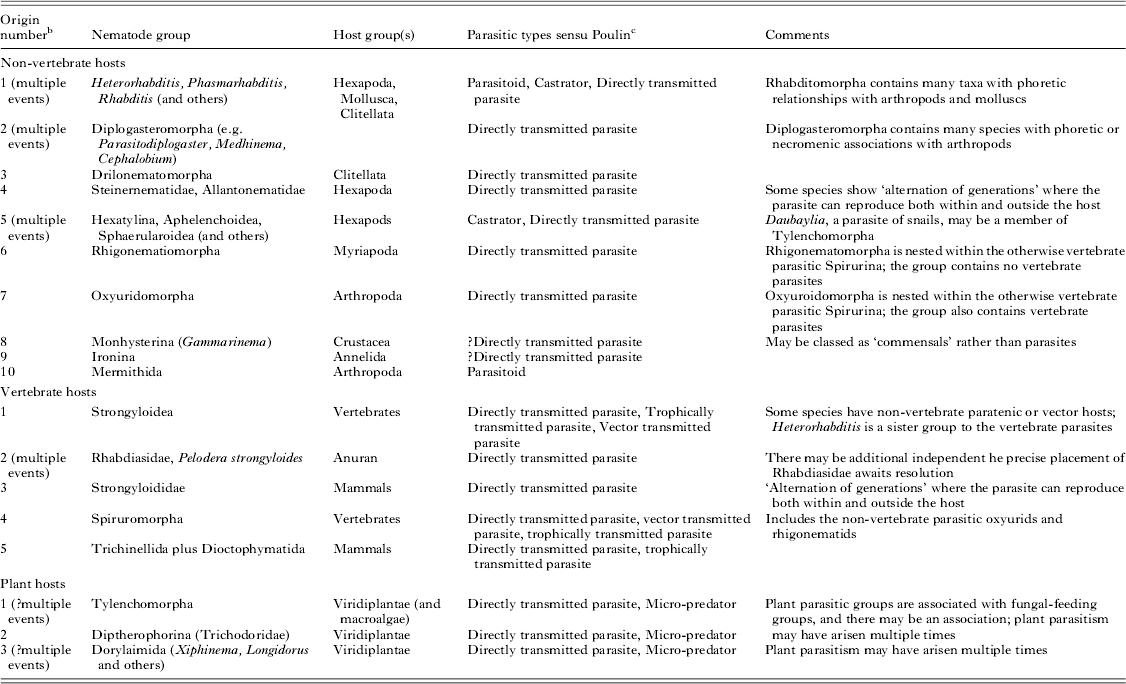
a There are many isolated additional descriptions of nematode associations with other taxa.
b The numbering of events follows Fig. 1.
c See Poulin (Reference Poulin2011); Poulin and Randhawa (Reference Poulin and Randhawa2013) for details.
There are some striking phylogenetic associations between non-vertebrate and vertebrate parasites. The Strongylomorpha (in Rhabditina), a major group of gut and airway parasites of vertebrates, are sisters to the Heterorhabditidae. Heterorhabditis species are entomopathogens that invade the haemocoel of insect larvae, and release a symbiotic bacterium that kills the host. Similarly, the mammal-parasitic Strongyloidoidae (Tylenchina; Panagrolaimomorpha) are related to the entomopathogenic Steinernematidae. Steinernema species also invade the haemocoel, and use a bacterial symbiont (a different group of species) to kill their hosts. The entomopathogens show convergence in life style, and are phylogenetically associated with a transition to vertebrate parasitism. However, there is no evidence that the vertebrate parasites utilize symbiotic bacteria during their life cycle. One model for the origin of the vertebrate-parasitic Strongylomorphs and Strongyloidoids is that they represent host capture by an ancestral terrestrial, entomopathogenic or entomoparasitic species, and subsequent radiation in the new host groups. Not all associations with arthropods necessarily lead to full parasitism, as there are several nematode groups (notably the Diplogasteromorpha in Rhabditina) where many species are phoretic or necromenic associates of arthropods but very few parasites have been found.
The Spirurina (Chromadoria; Rhabditida) are all parasites, mostly in vertebrates. Many Spirurina utilize vector hosts to facilitate transit from one definitive host to another, but there are also groups (Ascaridomorpha, Oxyruidomorpha) that do not use intermediate hosts. Some groups utilize multiple intermediate hosts, with Gnathostomatomorpha passaging through both a first, crustacean paratenic host and a second, fish host before establishing in carnivorous mammals. Surprisingly, in the new Spirurina phylogeny, groups with a simple, direct life cycle appear to be derived from a radiation of groups that have vector hosts. Gnathostomatomorpha arise basally, and Ascaridomorpha and Oxyuridomorpha have their origins as sisters to other vector-borne groups such as Spiruromorpha (Nadler et al. Reference Nadler, Carreno, Mejia-Madrid, Ullberg, Pagan, Houston and Hugot2007; Laetsch et al. Reference Laetsch, Heitlinger, Taraschewski, Nadler and Blaxter2012). Loss and gain of vector hosts within groups are common, and radical shift of vector species while continuing to use similar definitive hosts is a common feature of related parasites. For example, the Onchocercinae, parasitizing rodents, ungulates and primates, utilize mosquitoes, tabanid flies, black flies and mites as vectors. Two groups of arthropod parasites are nested within the otherwise vertebrate parasitic Spirurina (Rhigonematomorpha are gut parasites of large millipedes, and many Oxyuridomorpha are parasites of arthropods), and thus there must have been at least two independent transitions from vertebrate to arthropod parasitism (possibly by neotenic development in vector species) in these groups. Interestingly, Philometra obturans, a dracunculoid parasite of pike, has been observed to grow to sexual maturity in copepod intermediate hosts when the fish definitive host is absent (Moravec and de Buron, Reference Moravec and de Buron2013).
COMMON THEMES
There are few common themes in the parasitic lifestyles of nematodes. One is that the stage at which parasites transition from a free-living portion of the life cycle to the parasitic portion (or vice versa) is usually the J3/L3 stage, the same stage usually involved in phoretic associations (Sudhaus, Reference Sudhaus2010). Most rhabditid parasites first invade their hosts as J3, and mermithids exit their hosts as J3. The diapausing J3 stage of the free-living nematode Caenorhabditis elegans (not a parasite, but a species that has phoretic associations with terrestrial isopods) is particularly resistant to environmental insult, and is used as a model for the infective J3 of parasitic nematodes (particularly in Rhabditomorpha). It has been suggested that adaptation to rich food sources low in available oxygen such as rotting vegetation might predispose some groups to the acquisition of parasitic habits as the guts of other animals might offer a similar set of environmental challenges.
Echoes of the ‘great chain of being’, a world view common since pre-Darwinian times but still expressed today, can be heard in many discussions of parasite evolution. Parasites are suggested to be likely to show regression, losing morphological and metabolic complexity as they come to rely on hosts for more and more of the details of life as a metazoan. However, while a gut parasite might rely on the host for metabolic provisioning (thus implying a scope for simplification), it may also be subject to new stresses and demands in terms of countering host immunity (implying evolution of novelty). Similarly, parasites that utilize multiple hosts, or have major transitions in lifestyle, might be expected to gain or at least retain metabolic complexity, as they require toolkits to thrive in several distinct environments. Morphological novelties are common in parasitic nematodes: buccal teeth in hookworms (Chromadoria; Strongylomorpha); tagmatization in Trichuris (Dorylaimia; Trichinellidae); ornate spines, flanges and other cuticular decorations observed in arthropod-parasitic Oxyuridomorpha and Rhigonematomorpha (Chromadoria; Spirurina).
Even parasites with apparently simple life cycles can have very complex biology. Most Ascaridids infect new hosts through ingestion of embryonated eggs from the environment, and are often used as examples of simple, direct life cycles. Ascaris suum, which enters the host intestine as larvae arrested within a protective eggshell, but then invades the gut wall, enters the bloodstream, and reinfects the gut by exiting from the capillaries in the lungs, climbing the tracheal tree and returning to the gut by being swallowed, has a very complex in-host life cycle. This migration seems against reason, as it is costly to the parasite, and has been argued to be retention of an ancient trait of migration derived from a vector-borne ancestor. Retention of a costly phenotype in contemporary species requires explanation. The answer, derived by analysis of species pairs that differ in whether they migrate or not, is simply that migration is strongly associated with adult body size and with adult fecundity: migration is an adaptive trait maintained by selection (Skorping et al. Reference Skorping, Read and Keymer1991; Read and Skorping, Reference Read, Skorping, Griffin, Gwynn and Masson1995a , Reference Read and Skorping b ). It may be that the ability to migrate was inherited from an ancestral species, but its presence in living taxa is because it is currently adaptive.
GENOMICS AND PARASITISM
The first metazoan genome to be sequenced was that of a nematode: C. elegans (Chromadoria; Rhabditomorpha) (The C. elegans Genome Sequencing Consortium 1998) and nematode parasitologists were early adopters of the genomics toolkit (Blaxter et al. Reference Blaxter, Raghavan, Ghosh, Guiliano, Lu, Williams, Slatko and Scott1996, Reference Blaxter, Daub, Waterfall, Guiliano, Williams, Jayaraman, Ramzy, Slatko, Scott, Abad, Burnell, Laumond, Boemare and Coudert1997). A major effort over the past 20 years has delivered a large body of data on the transcribed genes of nematodes, initially through Sanger dideoxy sequencing expressed sequence tag sampling approaches (Parkinson et al. Reference Parkinson, Mitreva, Whitton, Thomson, Daub, Martin, Schmid, Hall, Barrell, Waterston, McCarter and Blaxter2004) but latterly using the new sequencing technologies to deliver whole transcriptomes (Blaxter et al. Reference Blaxter, Kumar, Kaur, Koutsovoulos and Elsworth2012). The roster of nematode species with significant transcriptome data publicly available now exceeds 100. These data have been critical in revealing genes associated with parasitic traits, and in identifying new drug targets and vaccine candidates (Elsworth et al. Reference Elsworth, Wasmuth and Blaxter2011).
The first parasitic nematode genome to be sequenced was that of Brugia malayi (Chromadoria; Spiruromorpha) (Ghedin et al. Reference Ghedin, Wang, Spiro, Caler, Zhao, Crabtree, Allen, Delcher, Guiliano, Miranda-Saavedra, Angiuoli, Creasy, Amedeo, Haas, El-Sayed, Wortman, Feldblyum, Tallon, Schatz, Shumway, Koo, Salzberg, Schobel, Pertea, Pop, White, Barton, Carlow, Crawford, Daub, Dimmic, Estes, Foster, Ganatra, Gregory, Johnson, Jin, Komuniecki, Korf, Kumar, Laney, Li, Li, Lindblom, Lustigman, Ma, Maina, Martin, McCarter, McReynolds, Mitreva, Nutman, Parkinson, Peregrin-Alvarez, Poole, Ren, Saunders, Sluder, Smith, Stanke, Unnasch, Ware, Wei, Weil, Williams, Zhang, Williams, Fraser-Liggett, Slatko, Blaxter and Scott2007). To date 19 nematode genome sequences have been published (see Fig. 1A and Table 2), and the majority of these genomes derive from parasites. As might be expected there is a bias in the species chosen for sequencing, and vertebrate and especially human parasites are over-represented. New sequencing technologies allow a new draft genome to be generated with very little resource, and more and more groups are now exploring the genomes of their target taxa. A loose alliance of nematode genome-interested biologists has been founded, and is nucleating the sequencing of as many genomes across the diversity of the phylum as is possible. The 959 Nematode Genomes initiative (Kumar et al. Reference Kumar, Koutsovoulos, Kaur and Blaxter2012a , Reference Kumar, Schiffer and Blaxter b ) mimics other community genome programmes such as the insect 5k project (i5K Consortium, 2013) (excepting that the headline goal is derived from the number of somatic cells in the adult female hermaphrodite C. elegans). It allows contributors to post information on the genomes they are pursuing and update the community as to their successes, and is a clearing-house for queries and collaborations (see http://www.nematodegenomes.org). By showing that genome sequencing, assembly, annotation and interpretation are within the reach of any research community, the initiative now has recorded well over 100 species for which genome data have been or are being generated.
Table 2. Nematode genome sequences
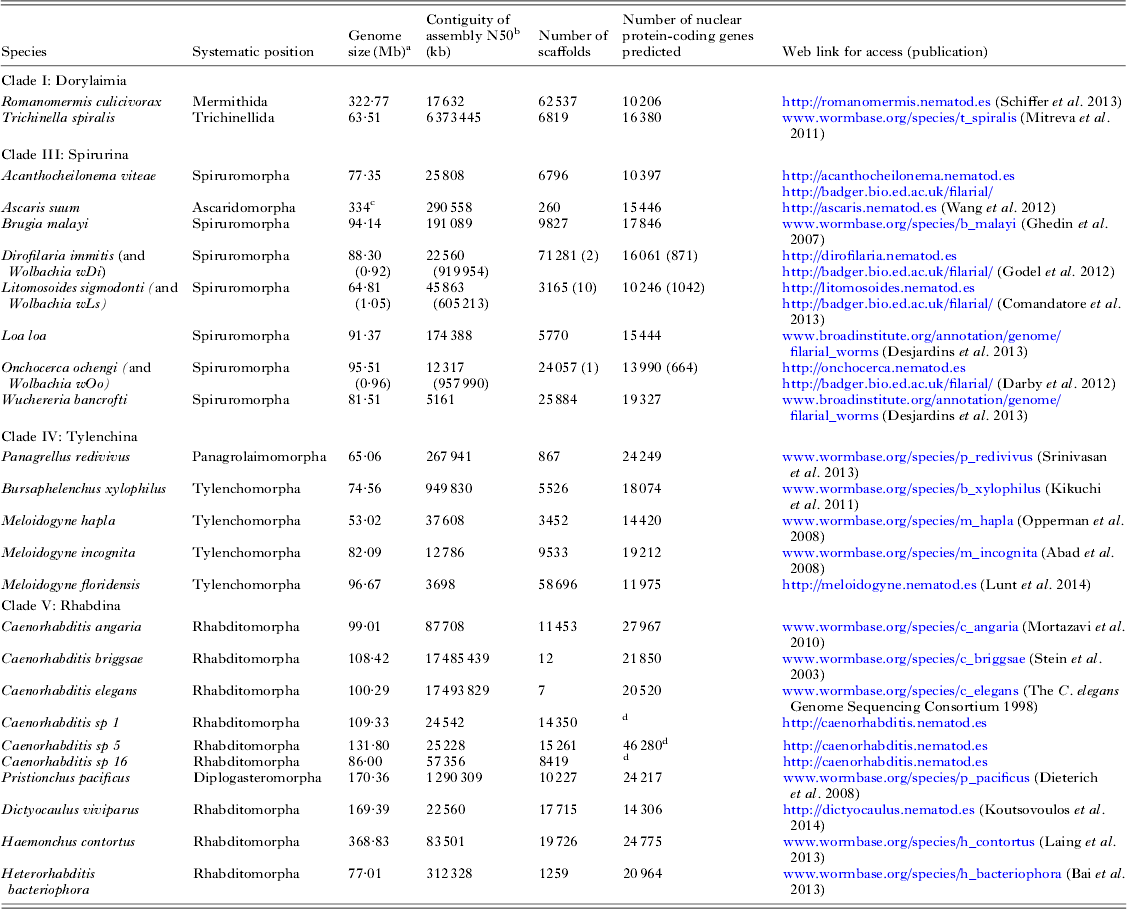
a Genome size is estimated from the span of genome assembly.
b The N50 is the weighted median scaffold length (the scaffold length at which 50% of the assembled genome is in scaffold of that length or greater).
c The A. suum genome undergoes chromatin diminution such that the somatic genome is ~40 Mb smaller than the germline genome (Wang et al. Reference Wang, Mitreva, Berriman, Thorne, Magrini, Koutsovoulos, Kumar, Blaxter and Davis2012).
d The gene predictions for Caenorhabditis sp. 5 are preliminary. The gene sets for the Caenorhabditis species are being re-predicted as part of a co-analysis across 10 Caenorhabditis genomes (E. Schwarz, M. Blaxter, unpublished).
We have been working to deliver genomic data for a number of species of key interest. These include free-living species (for example representatives of the under-sampled Enoplia, and additional Caenorhabditis species that will illuminate the biology of the model C. elegans) as well as parasites (Table 2). Using short-read next-generation sequencing technologies, and freely available assembly and annotation toolkits, we have been able to deliver important new genomes to the research communities. While generation of genomes of the quality and completeness of C. elegans would still require very significant effort, producing an assembly that is verifiably near-complete (albeit fragmented), and a gene set that is useful for a wide range of subsequent analyses, is relatively routine. A major remaining issue is that of heterozygosity in wild-sourced specimens. The C. elegans genome was derived from an essentially homozygous matrilineal clone, and the B. malayi genome from a highly inbred stock. Wild-caught, or recently colonized, nematode species will carry high levels of heterozygosity, and several animal and plant-parasitic nematode genome projects have struggled with the issues that extreme levels of heterozygosity (even in parasite lines maintained for a long time in laboratories) bring. Thus assemblies of new species may not reach the contiguity and completeness of the model genomes because of fundamental biological issues, and technical improvements and innovations, both wet laboratory and bioinformatic, are required.
Genome complexity does not appear to be closely tied to life habit. In terms of genome sizes, while some parasites (such as Meloidogyne hapla, with an estimated genome span of 54 Mb (Opperman et al. Reference Opperman, Bird, Williamson, Rokhsar, Burke, Cohn, Cromer, Diener, Gajan, Graham, Houfek, Liu, Mitros, Schaff, Schaffer, Scholl, Sosinski, Thomas and Windham2008)) have smaller genomes than those of free-living relatives (both cephalobes and rhabditids have genomes from 100–200 Mb), the largest genomes known thus far are in parasites (Romanomermis culicivorax has a genome of ~322 Mb (Schiffer et al. Reference Schiffer, Kroiher, Kraus, Koutsovoulos, Kumar, Camps, Nsah, Stappert, Morris, Heger, Altmuller, Frommolt, Nurnberg, Thomas, Blaxter and Schierenberg2013), A. suum 334 Mb (Wang et al. Reference Wang, Mitreva, Berriman, Thorne, Magrini, Koutsovoulos, Kumar, Blaxter and Davis2012) and Haemonchus contortus 370 Mb (Laing et al. Reference Laing, Kikuchi, Martinelli, Tsai, Beech, Redman, Holroyd, Bartley, Beasley, Britton, Curran, Devaney, Gilabert, Hunt, Jackson, Johnston, Kryukov, Li, Morrison, Reid, Sargison, Saunders, Wasmuth, Wolstenholme, Berriman, Gilleard and Cotton2013)). Within clades, genome sizes can vary widely (for example within the genus Meloidogyne sizes range from 54 Mb in M. hapla to 150 Mb estimated in Meloidogyne incognita (Abad et al. Reference Abad, Gouzy, Aury, Castagnone-Sereno, Danchin, Deleury, Perfus-Barbeoch, Anthouard, Artiguenave, Blok, Caillaud, Coutinho, Dasilva, De Luca, Deau, Esquibet, Flutre, Goldstone, Hamamouch, Hewezi, Jaillon, Jubin, Leonetti, Magliano, Maier, Markov, McVeigh, Pesole, Poulain, Robinson-Rechavi, Sallet, Segurens, Steinbach, Tytgat, Ugarte, van Ghelder, Veronico, Baum, Blaxter, Bleve-Zacheo, Davis, Ewbank, Favery, Grenier, Henrissat, Jones, Laudet, Maule, Quesneville, Rosso, Schiex, Smant, Weissenbach and Wincker2008; Lunt et al. Reference Lunt, Kumar, Koutsovoulos and Blaxter2014)), and this is associated with polyploidy. The knowledge of genome sizes of free-living nematodes outside the Rhabditomorpha is very sparse, and it may be that free-living, diploid species’ genomes regularly exceed the two-fold range known thus far. In general, the numbers of genes predicted from parasitic species are less than or equivalent to those predicted from free-living species. Caenorhabditis elegans has ~21 800 protein-coding genes, while H. contortus (Chromadoria; Strongylomorpha) has 20 600 (Laing et al. Reference Laing, Kikuchi, Martinelli, Tsai, Beech, Redman, Holroyd, Bartley, Beasley, Britton, Curran, Devaney, Gilabert, Hunt, Jackson, Johnston, Kryukov, Li, Morrison, Reid, Sargison, Saunders, Wasmuth, Wolstenholme, Berriman, Gilleard and Cotton2013). However, the mermithid R. culicivorax (Dorylaimia; Mermithida) has ~12 000 (Schiffer et al. Reference Schiffer, Kroiher, Kraus, Koutsovoulos, Kumar, Camps, Nsah, Stappert, Morris, Heger, Altmuller, Frommolt, Nurnberg, Thomas, Blaxter and Schierenberg2013), Trichinella spiralis (Dorylaimia; Trichinellida) has only 15 800 (Mitreva et al. Reference Mitreva, Jasmer, Zarlenga, Wang, Abubucker, Martin, Taylor, Yin, Fulton, Minx, Yang, Warren, Fulton, Bhonagiri, Zhang, Hallsworth-Pepin, Clifton, McCarter, Appleton, Mardis and Wilson2011), and the onchocercid nematodes (B. malayi and relatives; Chromadoria; Spiruromorpha) have between 12 000 and 14 000 (Godel et al. Reference Godel, Kumar, Koutsovoulos, Ludin, Nilsson, Comandatore, Wrobel, Thompson, Schmid, Goto, Bringaud, Wolstenholme, Bandi, Epe, Kaminsky, Blaxter and Maser2012; Desjardins et al. Reference Desjardins, Cerqueira, Goldberg, Dunning Hotopp, Haas, Zucker, Ribeiro, Saif, Levin, Fan, Zeng, Russ, Wortman, Fink, Birren and Nutman2013). Whether this reveals a loss of genic complexity in some parasites, or an overall more complex genomic heritage in the Rhabditina remains unclear. Changes in genome size are generally reflected in congruent changes in intron length, intergenic distance and repeat content.
HORIZONTAL GENE TRANSFERS INTO NEMATODE GENOMES
Plant parasitic nematodes must overcome the formidable defences of the plant cell wall in order to extract nutrition from their hosts. In addition, many sedentary plant parasites induce galls of various forms, structures induced in the plant by a parasite that must ‘know’ some of the tricks of plant developmental biology. Plant parasitic species secrete cellulolytic enzymes, and cloning and sequencing of these effectors revealed that some had their closest homologues in rhizosphere fungi and bacteria rather than other animals (Smant et al. Reference Smant, Stokkermans, Yan, de Boer, Baum, Wang, Hussey, Gommers, Henrissat, Davis, Helder, Schots and Bakker1998). These enzymes became the first robustly supported instances of horizontal or horizontal gene transfer into nematode genomes. A diverse roster of plant cell wall-degrading enzymes is now known from cyst and root-knot nematodes (Bird et al. Reference Bird, Jones, Opperman, Kikuchi and Danchin2014), and supplemented by piecemeal understanding of the repertoires of other phytopathogenic species. Phylogenetic inference suggests that these horizontally transferred genes were acquired early in the evolution of the Tylenchomorpha (Danchin et al. Reference Danchin, Rosso, Vieira, de Almeida-Engler, Coutinho, Henrissat and Abad2010; Rybarczyk-Mydlowska et al. Reference Rybarczyk-Mydlowska, Maboreke, van Megen, van den Elsen, Mooyman, Smant, Bakker and Helder2012). It is likely, given the inferred species of origin of the genes, that there were several independent events of gene acquisition (Mitreva et al. Reference Mitreva, Smant and Helder2009). Surveying tylenchomorph transcriptomes and genomes for genes with sequence-similarity profiles similar to those of the validated horizontal gene transfers reveals a wide range of candidates that include nod factor homologues and genes of unknown function that have disjunct distributions in plants and plant-parasitic nematodes, or in root bacteria and plant parasitic nematodes (Elsworth et al. Reference Elsworth, Wasmuth and Blaxter2011).
Importantly, horizontally transferred plant cell wall-degrading enzymes have also been identified in parasites from the other independently evolved plant–parasitic groups (Diptherophorina in Enoplia and Dorylaimida in Dorylaimia), suggesting that while the transition to phytophagy was difficult, it involved similar evolutionary trajectories in each case: the acquisition of the necessary toolkit from professional saprophytes in the root environment. It is interesting in this respect that similar cellulolytic enzymes have been identified in the free-living Pristionchus pacificus and related species (Rhabditina; Diplogateromorpha) (Mayer et al. Reference Mayer, Schuster, Bartelmes, Dieterich and Sommer2011). Horizontally acquired genes are evident in other species, including C. elegans (Parkinson and Blaxter, Reference Parkinson and Blaxter2003).
The close association between animal parasites and their hosts, and the requirement to specifically modulate especially the adaptive and anamnestic immune responses of vertebrate hosts, raises the question as to whether animal parasites also acquired novel genes from their hosts, or other commensals of these hosts. Surveys of expressed sequence tag datasets from plant parasitic species identified many potential horizontal transfer candidates, but very few candidates were identified in either transcriptome data from across the diversity of animal parasites, or in the genomes of animal parasites in Strongylomorpha, Ascaridomorpha, Spiruromorpha or Trichinellida (Elsworth et al. Reference Elsworth, Wasmuth and Blaxter2011). One interesting horizontally acquired gene in the Spiruromorpha is an alphaproteobacteria-like, second copy of ferrochelatase, an enzyme involved in the synthesis of haem (Elsworth et al. Reference Elsworth, Wasmuth and Blaxter2011). This gene is present in onchocercid nematodes (B. malayi and relatives), and suggests an additional or distinct requirement for haem in these tissue and blood parasites (Wu et al. Reference Wu, Novelli, Jiang, Dailey, Landmann, Ford, Taylor, Carlow, Kumar, Foster and Slatko2013). The ferrochelatase is quite distinct from that found in the Wolbachia symbionts of onchocercids (see below).
BACTERIAL SYMBIONTS
The long history of the tree of life is punctuated by many, highly significant events of symbiosis. In the Nematoda, several distinct types of symbiosis with bacteria have been recorded (Murfin et al. Reference Murfin, Dillman, Foster, Bulgheresi, Slatko, Sternberg and Goodrich-Blair2012). The free-living Stilbonematidae (Chromadoria) associate specifically with gammaproteobacteria that grow as a lawn on the nematodes’ cuticle. These sulphur-fixing bacteria act as a major food source for the nematode, and the nematodes ‘farm’ their bacterial associates by migrating to H2S-rich sediment layers (Bulgheresi Reference Bulgheresi2011; Murfin et al. Reference Murfin, Dillman, Foster, Bulgheresi, Slatko, Sternberg and Goodrich-Blair2012). In Anoplostoma (Enoplia), adult nematodes have neither mouth nor anus, and their guts are filled with a sulphur-fixing symbiont. Other similar trophic symbioses undoubtedly await discovery.
The entomopathogenic genera Heterorhabditis (Rhabditina, Strongylomorpha) (Bai et al. Reference Bai, Adams, Ciche, Clifton, Gaugler, Kim, Spieth, Sternberg, Wilson and Grewal2013) and Steinernema (Tylenchina, Panagrolaimomorpha) (Goodrich-Blair, Reference Goodrich-Blair2007) share a life-cycle strategy that utilizes specific Entobacteriaciae bacterial symbionts (Photorhabdus with Heterorhabditis, Xenorhabdus with Steinernema) to kill newly invaded insect larvae, and then to provide nutrition to the growing and reproducing nematodes. While the symbionts used and the details of the interactions differ, the convergence of these two nematode genera on the same general strategy is remarkable. In the plant-parasitic Xiphinema (Dorylaimia; Dorylaimida), a veruccomicrobial symbiont, Xiphinematobacter, is found intracellularly (Vandekerckhove et al. Reference Vandekerckhove, Willems, Gillis and Coomans2000). Its role in the biology of the nematode is largely unknown, although the symbiont is maternally transmitted and may play a role in modification of the nematodes’ reproductive mode.
The alphaproteobacterium Wolbachia pipientis was first described from insects, where they are reproductive parasites, manipulating the reproductive status, gender or sexual compatibility of their hosts (O'Neill, Reference O'Neill1995; Werren, Reference Werren1997). Wolbachia have subsequently been found in a range of terrestrial arthropods, and from nematodes. Molecular phylogenetic data suggest the presence of supergroups of Wolbachia that have distinct biology and host distributions (Lo et al. Reference Lo, Casiraghi, Salati, Bazzocchi and Bandi2002). Supergroup A and B Wolbachia are most widespread, and are found in insects. Nematodes are infected with supergroup C, D and F Wolbachia. Wolbachia have been described from the Spiruromorpha (in the Onchocercidae), and the Tylenchomorpha (Radopholus similis is the only species with infection described to date) (Jacob et al. Reference Jacob, Mitreva, Vanholme and Gheysen2008; Haegeman et al. Reference Haegeman, Vanholme, Jacob, Vandekerckhove, Claeys, Borgonie and Gheysen2009). General surveys using Wolbachia-specific PCR assays of many other nematode species have been negative (Bordenstein et al. Reference Bordenstein, Fitch and Werren2003; Duron and Gavotte, Reference Duron and Gavotte2007; Foster et al. Reference Foster, Kumar, Ford, Johnston, Ben, Graeff-Teixeira and Taylor2008). However, in R. similis (Tylenchina, Tylenchomorpha) the identification of expressed sequence tags corresponding to likely Wolbachia genes led to the identification of an intracellular symbiont in this plant parasite (Jacob et al. Reference Jacob, Mitreva, Vanholme and Gheysen2008; Haegeman et al. Reference Haegeman, Vanholme, Jacob, Vandekerckhove, Claeys, Borgonie and Gheysen2009).
In supergroups A and B, the symbiont phylogeny does not match that of its hosts, and host species tend to include both infected and uninfected individuals, reflecting frequent loss and acquisition of the symbiont through phylogenetic time. This pattern reflects the parasitic nature of the symbiosis. In contrast, supergroup C and D Wolbachia from onchocercid nematodes show traits suggestive of long, and possibly essential, mutualist interactions. The Wolbachia and nematode host phylogenies are congruent, indicating little if any host switching (Bandi et al. Reference Bandi, Anderson, Genchi and Blaxter1998; Casiraghi et al. Reference Casiraghi, Bain, Guerrero, Martin, Pocacqua, Gardner, Franceschi and Bandi2004). In infected species, all individuals are infected, and killing of the Wolbachia with antibiotics such as tetracyclines also affects the viability of the nematode host, with loss of fecundity and nematode death (Bandi et al. Reference Bandi, McCall, Genchi, Corona, Venco and Sacchi1999; Hoerauf et al. Reference Hoerauf, Nissen-Pahle, Schmetz, Henkle-Duhrsen, Blaxter, Buttner, Gallin, Al-Qaoud, Lucius and Fleischer1999; Landmann et al. Reference Landmann, Voronin, Sullivan and Taylor2011, Reference Landmann, Bain, Martin, Uni, Taylor and Sullivan2012). The exact nature of the mutualism remains unclear: the Wolbachia may assist the nematode metabolically (the distribution of bacteria in adult nematodes is reminiscent of the distribution of essential nutritive Buchnera bacteria in aphids) or in evading the vertebrate host's immune system (by confusing T-helper cell polarization with bacterial and metazoan signals at the same time) (Fenn and Blaxter, Reference Fenn and Blaxter2004, Reference Fenn and Blaxter2007; Darby et al. Reference Darby, Armstrong, Bah, Kaur, Hughes, Kay, Koldkjaer, Rainbow, Radford, Blaxter, Tanya, Trees, Cordaux, Wastling and Makepeace2012). Genome sequencing of filarial Wolbachia has permitted culling of the possible hypotheses for essentiality, but has not yielded data that definitively support specific metabolic vs immunoprotective roles (Darby et al. Reference Darby, Armstrong, Bah, Kaur, Hughes, Kay, Koldkjaer, Rainbow, Radford, Blaxter, Tanya, Trees, Cordaux, Wastling and Makepeace2012). It is also possible that the interference of the Wolbachia with oogenesis and development (Landmann et al. Reference Landmann, Voronin, Sullivan and Taylor2011, Reference Landmann, Bain, Martin, Uni, Taylor and Sullivan2012) makes it difficult, in evolutionary terms, for the nematode to rid itself of the symbionts. The symbiosis is not essential in the phylogenetic long term, as there are onchocercid species, such as Loa loa, Onchocerca flexuosa and Acanthocheilonema viteae, which have lost the infection and are now Wolbachia-free. Genomically, filarial Wolbachia display the expected phenotypes of mutualist endosymbionts: the genomes are reduced compared with the insect-parasitic supergroups A and B, with fewer protein coding genes and a lack of mobile elements such as phage (Comandatore et al. Reference Comandatore, Sassera, Montagna, Kumar, Koutsovoulos, Thomas, Repton, Babayan, Gray, Cordaux, Darby, Makepeace and Blaxter2013).
The onchocercid nematodes that lack living Wolbachia still retain a signature of past infection in the form of horizontally transferred fragments of Wolbachia-like DNA in their nuclear genomes (McNulty et al. Reference McNulty, Fischer, Curtis, Weil, Brattig and Fischer2013). Species that have live Wolbachia also have horizontally transferred Wolbachia-like fragments in their genomes (Dunning-Hotopp et al. Reference Dunning-Hotopp, Clark, Oliveira, Foster, Fischer, Munoz Torres, Giebel, Kumar, Ishmael, Wang, Ingram, Nene, Shepard, Tomkins, Richards, Spiro, Ghedin, Slatko, Tettelin and Werren2007; Ioannidis et al. Reference Ioannidis, Johnston, Riley, Kumar, White, Olarte, Ott, Tallon, Foster, Taylor and Dunning Hotopp2013). Horizontal transfer of organellar DNA to the nucleus is common, and thus the presence of these Wolbachia fragments could simply be a product of non-functional, stochastic incorporation of Wolbachia fragments into the genome (Blaxter, Reference Blaxter2007). More excitingly, these inserted fragments could be being used by the nuclear genome to express new, Wolbachia-derived proteins. While some Wolbachia fragments are expressed at low levels (Ioannidis et al. Reference Ioannidis, Johnston, Riley, Kumar, White, Olarte, Ott, Tallon, Foster, Taylor and Dunning Hotopp2013), most are not, and most are gene fragments that also have disabling mutations that render them inactive. Comparisons between the nuclear genomes of onchocercid species with and without Wolbachia has identified few shared insertions and no smoking gun of a constrained, conserved transfer that might substitute for a live Wolbachia.
The other Wolbachia found in nematodes are much less well-studied. Some onchocercid nematodes carry a Wolbachia that is placed in supergroup F, alongside Wolbachia from termites, fleas and bedbugs (Bordenstein et al. Reference Bordenstein, Fitch and Werren2003; Duron and Gavotte, Reference Duron and Gavotte2007; Foster et al. Reference Foster, Kumar, Ford, Johnston, Ben, Graeff-Teixeira and Taylor2008; Jacob et al. Reference Jacob, Mitreva, Vanholme and Gheysen2008; Haegeman et al. Reference Haegeman, Vanholme, Jacob, Vandekerckhove, Claeys, Borgonie and Gheysen2009; Comandatore et al. Reference Comandatore, Sassera, Montagna, Kumar, Koutsovoulos, Thomas, Repton, Babayan, Gray, Cordaux, Darby, Makepeace and Blaxter2013). Initial analyses suggested that the R. similis Wolbachia was distantly related to any other supergroup (Jacob et al. Reference Jacob, Mitreva, Vanholme and Gheysen2008; Haegeman et al. Reference Haegeman, Vanholme, Jacob, Vandekerckhove, Claeys, Borgonie and Gheysen2009), but this result is questionable (Koutsovoulos et al. Reference Koutsovoulos, Makepeace, Tanya and Blaxter2014). The bovine lungworm Dictyocaulus viviparus (Rhabditina, Strongylomorpha) was not known to have any association with Wolbachia until its genome was sequenced (Koutsovoulos et al. Reference Koutsovoulos, Makepeace, Tanya and Blaxter2014). Within the nuclear genome contigs were ~1 Mb of DNA fragments that had highest similarity to Wolbachia genomes (Koutsovoulos et al. Reference Koutsovoulos, Makepeace, Tanya and Blaxter2014). These fragments bore all the hallmarks of being non-functional horizontal transfers from a once-present Wolbachia. Using these horizontally transferred fragments, the likely source of the transfer was identified as a supergroup F Wolbachia. The D. viviparus data allow resolution of the relationship of supergroup F (and the Wolbachia from R. similis) as sisters to supergroups C and D. The fragments in the D. viviparus genome included remnants of bacteriophage, suggesting that the source genome might have been more like that of the parasites of supergroup A and B than the reduced C and D symbionts. It will be important to survey other emerging nematode genomes for evidence of past association with Wolbachia, and perhaps other bacteria, and thus reveal the extent of the interactions between these symbionts and nematodes, and perhaps even identify particular associations with parasitism.
A GENOME-BASED TREE OF NEMATODA
One critical issue that molecular phylogenetic analyses now face is that more data are needed. To date, most analyses have used a single locus, the nuclear small subunit ribosomal RNA gene (nSSU). The nSSU is a good gene for deep phylogenetics, but the phylogenetic history that can be extracted from its ~1800 bases is limited. There are now over 8000 nematode nSSU sequences in the public sequence databases (many fragmentary) from over 4000 nominal species. It is not possible to derive a robustly supported tree from this many sequences, and many internal nodes that were unresolved in the earliest analyses remain unresolved in the most recent ones (Holterman et al. Reference Holterman, van der Wurff, van den Elsen, van Megen, Bongers, Holovachov, Bakker and Helder2006; van Megen et al. Reference van Megen, van den Elsen, Holterman, Karssen, Mooyman, Bongers, Holovachov, Bakker and Helder2009; Bik et al. Reference Bik, Lambshead, Thomas and Lunt2010), probably because of a lack of unambiguous signal in the single, short nSSU locus. The mitochondrial genome is a readily accessed source of data for phylogenetic inference, and complete genomes are available for over 40 species. These have yielded phylogenies that are well-resolved but at odds with nSSU phylogenies (Park et al. Reference Park, Sultana, Lee, Kang, Kim, Min, Eom and Nadler2011; Sultana et al. Reference Sultana, Kim, Lee, Han, Kim, Min, Nadler and Park2013). Specifically, neither Spiruria (Blaxter et al. Reference Blaxter, De Ley, Garey, Liu, Scheldeman, Vierstraete, Vanfleteren, Mackey, Dorris, Frisse, Vida and Thomas1998) nor Tylenchina are recovered as monophyletic, and the sister relationship between Heterorhabditis bacteriophora and Strongylomorpha is not recovered (Park et al. Reference Park, Sultana, Lee, Kang, Kim, Min, Eom and Nadler2011; Sultana et al. Reference Sultana, Kim, Lee, Han, Kim, Min, Nadler and Park2013). Whether these differences arise from biases or errors in the nuclear or mitochondrial data that have not been mitigated remains to be clarified.
One key utility of the new genome-wide data from a wide range of nematode species is that we have a much larger set of data to draw on when compiling matrices for phylogenetic inference (Jones et al. Reference Jones, Koutsovoulos and Blaxter2011). A major issue is the selection of loci that are orthologous (i.e. where representatives in different species have their origin in a single instance in an ancestral species) and where data coverage is relatively complete. Using gene sets inferred from complete genome sequences, and also complete or high-density transcriptome data, it is possible to infer a set of orthologous genes across the breadth of the phylum. One approach to achieving this is to use a tool such as Core Eukaryotic Genes Mapping Approach (CEGMA) (Parra et al. Reference Parra, Bradnam and Korf2007), which identifies a set of 248 genes known to be present in six model eukaryote species. These genes tend to be highly conserved in sequence, and one limitation resulting from their use may be that there is not enough variation to record closely spaced, or recent branching patterns. An alternative approach is to generate a sequence-based clustering of all genes from all the species under study, using a tool such as orthoMCL (Li et al. Reference Li, Stoeckert and Roos2003), and to query the resultant data for putative sets of orthologues. Using these approaches it is possible to build datasets that include many hundreds of thousands of aligned nucleotides (and several hundred thousand aligned codons or amino acids). These datasets can then be used to address questions left unanswered by the nSSU datasets. First attempts to explore resolution of the deeper phylogeny of Nematoda with multiple nuclear genes derived from whole-genome sequencing priojects have largely supported the existing nSSU-derived phylogeny, albeit with limited taxon representation (Desjardins et al. Reference Desjardins, Cerqueira, Goldberg, Dunning Hotopp, Haas, Zucker, Ribeiro, Saif, Levin, Fan, Zeng, Russ, Wortman, Fink, Birren and Nutman2013; Laing et al. Reference Laing, Kikuchi, Martinelli, Tsai, Beech, Redman, Holroyd, Bartley, Beasley, Britton, Curran, Devaney, Gilabert, Hunt, Jackson, Johnston, Kryukov, Li, Morrison, Reid, Sargison, Saunders, Wasmuth, Wolstenholme, Berriman, Gilleard and Cotton2013).
The true power of this phylogenomic approach will only be realized when many hundred nematode genomes representing known diversity are sequenced, but already large datasets can be collated and explored. We have used a combination of whole-genome-derived and transcriptomics-derived gene sets to, firstly, attempt to replicate the findings from the nSSU analyses performed previously, and secondly to resolve some of the remaining unresolved polytomies and conflicts between analyses (Fig. 1B) (Blaxter et al. Reference Blaxter, Koutsovoulos, Jones, Kumar, Elsworth, Cotton, Hughes and Olson2014). Analyses were performed with data from 181 genes from 23 nematode taxa including representatives of the Dorylaimia, Enoplia and Chromadoria. Taxon sampling remained most limited in Enoplia (a single representative, Enoplus brevis), and in the comb-like series of ordinal taxa subtending the Rhabditida in Chromadoria (only Laxus oneistus from Chromadorida). With these taxa the major clades (I–V) that were identified using nSSU (Blaxter et al. Reference Blaxter, De Ley, Garey, Liu, Scheldeman, Vierstraete, Vanfleteren, Mackey, Dorris, Frisse, Vida and Thomas1998) were recovered, the branching order of clades III (Spirurina), IV (Tylenchina) and V (Rhabditina) was resolved as (III, (IV,V)), and the Enoplia were robustly resolved as arising basal to Dorylaimia plus Chromadoria. While E. brevis has a relatively short branch length in these analyses, we caution that its placement may be artefactual due to phylogenetic artefacts elsewhere in the tree, or outgroup problems.
PROSPECTS
The 959 Nematode Genomes initiative notes nearly 100 genomes in progress for the phylum. We have heard anecdotally of many more taxa where researchers are approaching their research goals through genome sequencing, or deep transcriptome sequencing. Improved sequencing technologies such as long single-molecule reads will improve the contiguity of genomes, and improved algorithms will enable assembly even in the presence of high levels of heterozygosity. Careful sampling, and methods for unbiased amplification of genomic DNA from single specimens will fill in the diversity of the tree, and multi-locus phylogenies provide deep resolution of relationships. The next few years will also see the development of rich collated resources for nematode genomes, including shared genome browsing environments, robust inferences of gene orthology and gene family evolution, and identification of genes and gene families that show particular patterns of evolution associated with distinct clades in the phylum. In addition, with the development of robust protocols for RNA-based interference in many species, and the development of specific genome editing methods that can be applied to any organism, we expect that questions of the specific roles of many genes to be elucidated. As ever, the questions remain biological: which traits and which genomic features are associated with parasitism, what selective forces maintain them, and how do these change through the ongoing struggle between host and parasite?
ACKNOWLEDGEMENTS
We thank our colleagues in Edinburgh and elsewhere who have been working with us on sequencing, assembly and annotation of nematode genomes, in particular Sujai Kumar. We thank Andrew Jackson, Matt Berriman and James Cotton for inviting MB to participate in the Wellcome Trust workshop on ‘The evolution of parasite genomes and the origins of parasitism’. We apologize to colleagues whose work we were unable to reference due to limitations of space.




 Open access
Open access