Chronic low-grade inflammation plays an important role in numerous diseases, including major depression and heart disease, and it has been implicated as one of the major causes for the comorbidity of these diseases (Capuron et al., Reference Capuron, Su, Miller, Bremner, Goldberg, Vogt and Vaccarino2008; Vaccarino et al., Reference Vaccarino, Johnson, Sheps, Reis, Kelsey and Bittner2007, Reference Vaccarino, Brennan, Miller, Bremner, Ritchie, Lindau and Hazen2008). The inflammatory response is activated by pro-inflammatory cytokines, of which Tumor Necrosis Factor-α (TNF-α) and Interleukin-1 (IL-1) are the first to appear (Tracey, Reference Tracey2002). The inflammatory cascade is further promoted by the production of IL-6 that in turn stimulates the acute-phase response, which is reflected in the synthesis of fibrinogen and C-Reactive Protein (CRP) (Gabay, Reference Gabay2006; Gabay & Kushner, Reference Gabay and Kushner1999; Packard & Libby, Reference Packard and Libby2008). Elevations in TNF-α, IL-6, CRP, and fibrinogen have been associated with an increased risk for both cardiac disease (Cesari et al., Reference Cesari, Penninx, Newman, Kritchevsky, Nicklas, Sutton-Tyrrell and Pahor2003; Danesh et al., Reference Danesh, Wheeler, Hirschfield, Eda, Eiriksdottir, Rumley and Gudnason2004, Reference Danesh, Kaptoge, Mann, Sarwar, Wood, Angleman and Gudnason2008; Humphries et al., Reference Humphries, Cooper, Talmud and Miller2007; Libby & Theroux, Reference Libby and Theroux2005; Packard & Libby, Reference Packard and Libby2008; Woods et al., Reference Woods, Brull, Humphries and Montgomery2000) and major depression (O'Brien et al., Reference O'Brien, Scott and Dinan2004; Penninx et al., Reference Penninx, Kritchevsky, Yaffe, Newman, Simonsick, Rubin and Pahor2003).
In spite of the obvious importance of these pro-inflammatory markers in depression and cardiovascular disease, both of which are in the top four of burden of disease prediction for 2020 (Mathers & Loncar, Reference Mathers and Loncar2006), very little is known about the etiology of individual differences in TNF-α, IL-6, CRP, and fibrinogen levels. An important first question is that to what extent the variance in these biological parameters is innate, caused by environmental factors that are shared by family members, or caused by environmental factors unique to each individual member of a family. This question can be addressed by the classical twin design comparing the resemblance between monozygotic (MZ) and dizygotic (DZ) twins (Boomsma et al., Reference Boomsma, Busjahn and Peltonen2002; van Dongen et al., Reference van Dongen, Slagboom, Draisma, Martin and Boomsma2012). A few twin studies in healthy samples have estimated the heritability of cytokines and acute phase reactants, with estimates varying between 21% and 60% for fibrinogen (de Lange et al., Reference de Lange, Snieder, Ariens, Spector and Grant2001, Reference de Lange, de Geus, Kluft, Meijer, van Doornen, Boomsma and Snieder2006; de Maat et al., Reference de Maat, Bladbjerg, Hjelmborg, Bathum, Jespersen and Christensen2004; Jermendy et al., Reference Jermendy, Horvath, Littvay, Steinbach, Jermendy, Tarnoki and Osztovits2011; Reed et al., Reference Reed, Tracy and Fabsitz1994; Su et al., Reference Su, Snieder, Miller, Ritchie, Bremner, Goldberg and Vaccarino2008), between 20% and 76% for CRP (de Maat et al., Reference de Maat, Bladbjerg, Hjelmborg, Bathum, Jespersen and Christensen2004; Jermendy et al., Reference Jermendy, Horvath, Littvay, Steinbach, Jermendy, Tarnoki and Osztovits2011; MacGregor et al., Reference MacGregor, Gallimore, Spector and Pepys2004; Rahman et al., Reference Rahman, Bennet, Pedersen, de, Svensson and Magnusson2009; Su et al., Reference Su, Snieder, Miller, Ritchie, Bremner, Goldberg and Vaccarino2008; Reference Su, Lampert, Zhao, Bremner, Miller, Snieder and Vaccarino2009a; Reference Su, Miller, Snieder, Bremner, Ritchie, Maisano and Vaccarino2009b; Wang et al., Reference Wang, Christoffel, Brickman, Hong, Arguelles, Zhang and Wang2011; Wessel et al., Reference Wessel, Moratorio, Rao, Mahata, Zhang, Greene and O'Connor2007; Worns et al., Reference Worns, Victor, Galle and Hohler2006), between 17% and 26% for TNF-α (de Maat et al., Reference de Maat, Bladbjerg, Hjelmborg, Bathum, Jespersen and Christensen2004; Sas et al., Reference Sas, Jamshidi, Zheng, Wu, Korf, Alizadeh and Snieder2012), and between 15% and 61% for IL-6 (de Maat et al., Reference de Maat, Bladbjerg, Hjelmborg, Bathum, Jespersen and Christensen2004; Grunnet et al., Reference Grunnet, Poulsen, Klarlund, Mandrup-Poulsen and Vaag2006; Sas et al., Reference Sas, Jamshidi, Zheng, Wu, Korf, Alizadeh and Snieder2012; Su et al., Reference Su, Snieder, Miller, Ritchie, Bremner, Goldberg and Vaccarino2008, Reference Su, Miller, Snieder, Bremner, Ritchie, Maisano and Vaccarino2009b, Reference Su, Lampert, Zhao, Bremner, Miller, Snieder and Vaccarino2009a; Worns et al., Reference Worns, Victor, Galle and Hohler2006). With a few exceptions, heritability estimates for the aforementioned studies have been based on relatively small twin samples. Such studies are fairly accurate in estimating broad-sense heritability, but they lack precision and power to estimate the contribution of non-additive genetic effects or shared family environment such as the dietary habits or neighborhood factors shared by parents and offspring. As the average sample size of previous studies was around 400 individuals, these studies were not sufficiently powered to detect an effect of shared environmental factors explaining less than 40% of the variance or to discriminate between additive and non-additive genetic factors (Posthuma & Boomsma, Reference Posthuma and Boomsma2000). Also, the relatively small sample sizes may explain the large range of heritability estimates based on previous studies.
Here we extend the classical twin design, including only MZ and DZ twin pairs, by including non-twin siblings and their parents in the largest set of twin- and family data on TNF-α, IL-6, CRP, and fibrinogen described to date. Inclusion of non-twin siblings increases statistical power and offers the possibility to assess twin-specific effects. The inclusion of parents allows the study to take into account assortative (non-random) mating effects, which can influence heritability estimates. Data from parents also allow for the examination of shared household effects in spouses who share a household, but are not biologically related (e.g., Distel et al., Reference Distel, Rebollo-Mesa, Abdellaoui, Derom, Willemsen, Cacioppo and Boomsma2010; Rebollo & Boomsma, Reference Rebollo and Boomsma2006). The availability of a large sample size allowed for the exclusion of subjects with current and recent illness and for the examination of a number of health-related variables and methodological factors that could affect the reliability of the assessment of plasma levels of inflammatory variables while retaining adequate power to detect shared environmental factors and to discriminate between additive and non-additive genetic factors.
Methods
Subjects
The data were obtained from the Netherlands Twin Register (NTR) Biobank study that was conducted among twins and their family members registered with the NTR during 2004–2008 (Willemsen et al., Reference Willemsen, de Geus, Bartels, van Beijsterveldt, Brooks, Estourgie-van Burk and Boomsma2010). Subjects were visited between 7 a.m. and 10 a.m. at home or, when preferred, at work, to collect blood and urine samples. Subjects were instructed to fast from the evening before, to abstain from physical exertion, and, if possible, not to take medication on the day of the home visit, and to refrain from smoking one hour before the home visit. Fertile women were visited on the 2nd–4th day of their menstrual cycle, or if they took oral contraceptives, during their pill-free week. During the visit, a brief interview was conducted on health status, including an inventory of medication use, illness (last time occurrence, duration and type of illness), and adherence to the protocol.
The study consisted of 9,405 subjects with data on at least one of the four pro-inflammatory parameters of interest. Values exceeding 15 pg/mL for IL-6 and TNF-α, 15 mg/L for CRP and/or 6 g/L for fibrinogen were set to be missing, leading to the exclusion of 11 subjects. Subjects who were on anti-inflammatory medication, medication impacting on the hypothalamic-pituitary-adrenal (HPA) axis, or both, were excluded from further analyses (N = 408). We also excluded subjects suffering from cold, flu, inflammation, or allergy at the time of blood sampling (N = 1,013). The remaining subjects (N = 7,973) served as the reference group to quantify the effects of various covariates and to compute residual scores for every immune parameter.
For the twin-family analyses we, in addition, excluded non-biological parents and siblings (N = 35), spouses of twins (N = 409), subjects under 18 years of age (N = 87), the third member of triplets, and additional twins from families with more than one twin pair (N = 4). When zygosity was missing for a twin pair and both twins participated in the study, we randomly selected one of the two to be excluded (N = 10). To simplify the genetic model fitting procedure, we included a maximum of two singleton brothers and two singleton sisters per family and randomly selected two siblings from families with more than two same-sex siblings (N = 99 excluded). The final sample comprised 3,095 families with 7,329 family members, of which 3,534 subjects were twins: more specifically, 590 MZ male (MZM), 320 DZ male (DZM), 1,281 MZ female (MZF), 624 DZ female (DZF), and 719 dizygotic opposite-sex (DOS) twins. The following complete twin pairs were included: 201 MZM, 96 DZM, 466 MZF, 211 DZF and 217 DOS. Furthermore, 464 non-twin male siblings, 1,104 non-twin female siblings, 1,003 fathers of twins, and 1,224 mothers of twins were included. Zygosity of twins was determined by DNA typing for 85.1% of the same-sex twin pairs. For the other same-sex pairs, zygosity was based on survey questions on physical similarity and the frequency of confusion of the twins by parents, other family members, and strangers. Agreement between the zygosity based on these items and the zygosity based on DNA was 96.1% (Willemsen et al., Reference Willemsen, Vink, Abdellaoui, den, van Beek, Draisma and Boomsma2013).
Assessment of TNF-α, IL-6, CRP, and Fibrinogen
During the home visit, eight blood tubes were collected in the following order: 2 × 9-mL EDTA, 2 × 9-mL heparin, 1 × 4.5-mL CTAD, 1 × 2-mL EDTA, and 1 × 4.5-mL serum. To prevent clotting, all tubes were inverted gently 8–10 times immediately after collection (for details, see Willemsen et al., Reference Willemsen, de Geus, Bartels, van Beijsterveldt, Brooks, Estourgie-van Burk and Boomsma2010).
TNF-α and IL-6 were measured in EDTA plasma obtained from one of the 9-mL tubes. During transportation this tube was stored in melting ice and upon arrival at the laboratory it was centrifuged for 20 min at 2000× g at 4°C. EDTA plasma, buffy coat, and red blood cells were harvested and aliquoted (0.5 mL), snap-frozen in dry ice, and stored at −30°C. Plasma levels of TNF-α and IL-6 were determined using an UltraSensitive ELISA (R&D systems, Minneapolis, USA, Quantikine HS HSTA00C). The inter-assay coefficient of variation (CV) for TNF-α was <12.8%, and for IL-6 this was <11.6%.
C-reactive protein was obtained from one of the 9-mL heparin tubes. The tube was stored in melting ice during transportation. At the laboratory, the tube was centrifugated for 15 min at 1000× g at 4°C, after which heparin plasma was obtained and divided into eight subsamples of 0.5 mL, snap-frozen, and stored at −30°C. The processing took place in a sterile flow cabinet. CRP level in heparin plasma was determined using the Immulite 1000 CRP assay (Diagnostic Product Corporation, USA). The inter-assay CV was <5.1%.
Fibrinogen level was obtained from the 4.5-mL CTAD tube, which was stored in melting ice during transportation. Upon arrival at the laboratory, it was centrifuged for 20 min at 2000× g at 4°C, after which citrated plasma was harvested from the buffy coat and red blood cells, aliquoted (0.5 mL), snap-frozen in dry ice, and stored at −30°C. Fibrinogen levels in CTAD plasma were determined on a STA Compact Analyzer (Diagnostica Stago, France), using STA Fibrinogen (Diagnostica Stago, France). The inter-assay CV was <6.1%.
Fibrinogen values were normally distributed whereas data on the other variables were skewed. Therefore, we took the natural logarithm of these values.
Assessment of Covariates
For the heritability analyses, we took into account the effects of age, sex, health-related covariates known to be associated with inflammatory parameters (body mass index [BMI], smoking status), and several methodological covariates that could lead to inflation of family correlations (month of blood sampling and batch effect). During the home visit, height and weight were assessed and BMI was calculated. Subjects were also asked about their past and current smoking behavior and were categorized into one of the following five groups: never smoked, ex-occasional smoker, ex-regular smoker, current occasional smoker, and current regular smoker. The month of blood sampling was used to correct for the effects of time of the year on the four pro-inflammatory markers. For the cytokines, we also took into account differences in values because of the plate on which the samples were processed by using the plate mean value for the cytokines as a covariate. The levels of the acute phase reactants were determined on a per sample basis, so plate effects for these variables are not applicable. Previous research suggested that when using the ELISA of R&D systems, individuals with blood group O may show higher TNF-α and IL-6 levels than other ABO blood groups, which may in part be due to assay-specific cross-reactivity with ABO antigens (Melzer et al., Reference Melzer, Perry, Hernandez, Corsi, Stevens, Rafferty and Ferrucci2008; Naitza et al., Reference Naitza, Porcu, Steri, Taub, Mulas, Xiao and Cucca2012). To investigate this potential confounding effect, of all the SNPs in the ABO gene region plus/minus a 10-Kb border, we used rs644234 SNP that showed the strongest association with TNF-α and IL-6 in our data. The rs644234 SNP explained 7% and 4% of TNF-α and IL-6 values, respectively. Data on this SNP were available for 5,950 healthy subjects with TNF-α data and for 5,947 subjects with IL-6 data. Because the twin-family models yielded similar results with and without taking the effect of the ABO SNP into account, we only report the analyses on the full sample.
Statistical Analyses
Data preparation, sample selection, and tests for the effects of covariates were conducted using IBM Statistical Package of Social Sciences 20.0. The covariates were included in a multiple regression analysis (forced entry) and the residual scores were saved for the heritability analyses. As there was a significant age-by-sex interaction for CRP, fibrinogen, and IL-6, regression coefficients for age were estimated separately for men and women for these variables. Genetic models were fitted to the data using structural equation modeling (SEM) in the software package Mx (Neale et al., Reference Neale, Boker, Xie and Maes2006). First, a fully parameterized, or saturated, model was fitted and a goodness-of-fit statistic based on minus twice the logarithm of the likelihood (-2LL) was calculated. Next, the fully saturated model was simplified to a more restricted model to test whether constraints were allowed to be put on the data. The comparison of fit of a restricted model to the full model is performed by means of likelihood-ratio (χ2) tests in which the difference in -2LL between the two models is calculated. When the likelihood-ratio test is significant (p < .01), the nested model is considered to fit significantly worse to the data than the fuller model it is tested against.
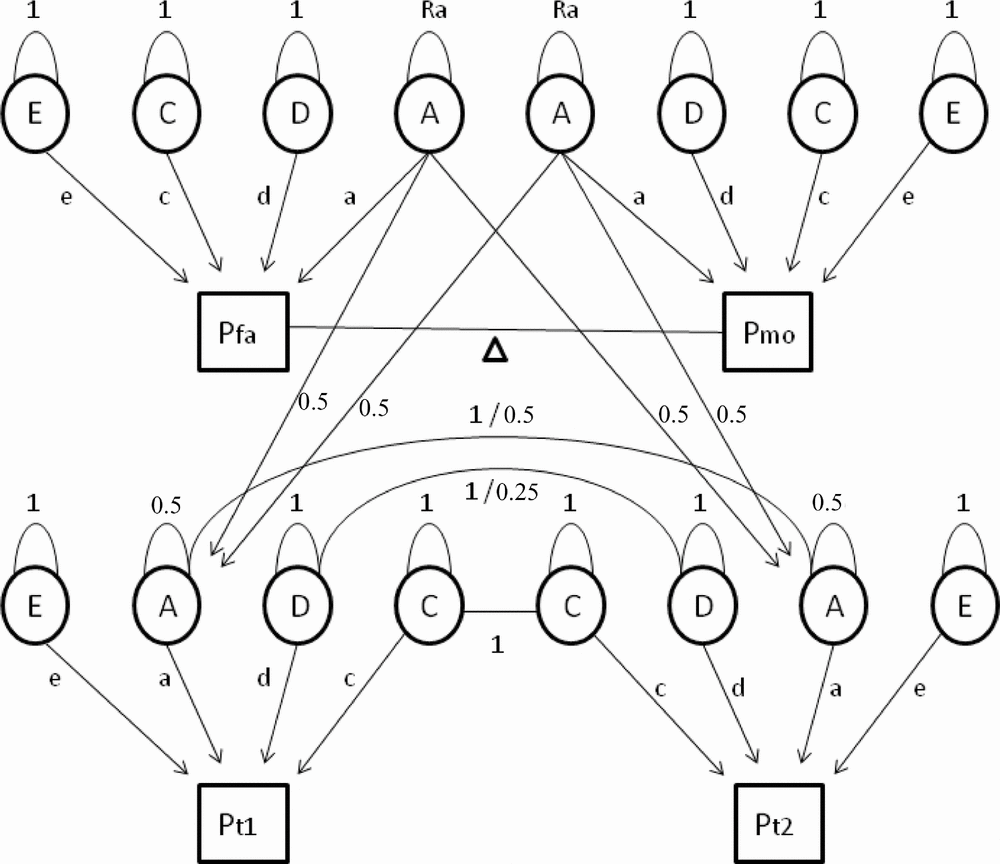
First, we tested whether constraints on the means and variances for men and women were allowed, and whether different types of family correlations were equal. In the full model, 19 parameters were estimated: separate means, standard deviations, and regression coefficients for age on the phenotype for men and women, and 13 family correlations (for MZM, MZF, DZM, DZF, DOS twin pairs, male siblings, female siblings, opposite-sex siblings and for mother–daughter, mother–son, father–daughter, father–son, and one spouse correlation). Quantitative sex differences, indicating that the heritability of a trait is different in men and women, were assessed by testing whether correlations in male–male and female–female pairs of first-degree relatives (DZ twins and non-twin siblings) were equal. Next, we tested whether the same genes regulate cytokine and acute phase reactant levels in men and women (Vink et al., Reference Vink, Bartels, van Beijsterveldt, van, van Beek, Distel and Boomsma2012). When the correlations for a trait are the same in the same-sex and opposite-sex pairs of family members, there is no evidence for qualitative sex differences in the genetic architecture. When the correlations in DZ twin pairs are of similar magnitude as the correlations in sib–sib pairs, there is no evidence for twin-specific resemblance. Generation effects were tested by equating parent–offspring correlations with the correlations between all other first-degree relatives (DZ twins and non-twin siblings). If this constraint is allowed, then there is no evidence that gene expression changes with age. Spousal resemblance was assessed by testing if the correlation between the parents of the twins was significantly different from zero. The most parsimonious model with the maximal number of allowable restrictions was carried forward to the genetic structural equation analyses. In these analyses, the family covariance structure is used to estimate the relative contribution of latent additive (A) and non-additive or dominant (D) genetic factors and common (C) and unique (E) environmental factors to the phenotypic variance. Based on the variance estimates from the full genetic model, sequentially constrained submodels were compared with the fit of the full model to arrive at the most parsimonious genetic model describing the total phenotypic variance best (see Figure 1 for a schematic representation of the extended twin-family model).

FIGURE 1 Path diagram of an extended twin family showing four subjects belonging to one family: father (Pfa), mother (Pmo), twin 1 (Pt1), and twin 2 (Pt2), where P is phenotype. In the genetic model (without constraints) a maximum of 10 parameters are estimated. Path loadings a, d, c, and e represent the influence of additive and non-additive genetic effects and shared and unique environmental factors on the phenotype. Spousal resemblance is modeled as a function of phenotypic assortment, denoted by the delta path (Δ). When the delta path coefficient is larger than zero, this indicates non-random mating. The residual variance of additive genetic effects in parents (Ra) will then become larger than 1 to account for increased transmission of additive genetic effects from parents to children under conditions of non-random mating. Finally, two means and two age regression coefficients (for males and females separately) are estimated. Fixed parameters include the 0.5 genetic relatedness of parents and their children (path running from A of both parents to A of their children with a path loading of 0.5). The residual variance of A in the offspring is constrained at 0.5. Finally, MZ twins correlate 1 regarding A and D factor scores (because they are assumed to share all of their genetic material), whereas DZ or sibling pairs correlate 0.5 and 0.25, respectively.
Results
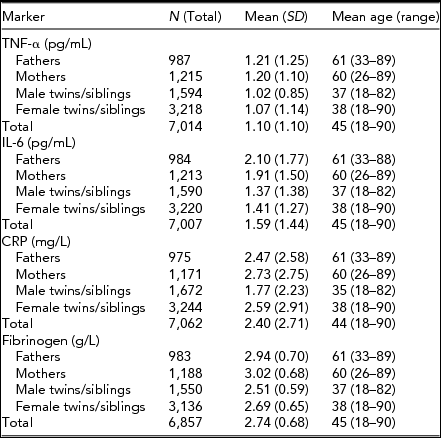
Descriptive statistics for the four immune parameters of interest in twins, siblings, and parents are given in Table 1. Table 2 presents the amount of variance explained by various technical and biological covariates that were taken into account.
TABLE 1 Mean Values (and Standard Deviations) and Mean Age (Range) for Tumor Necrosis Factor-α (TNF-α), Interleukin-6 (IL-6), C-Reactive Protein (CRP), and Fibrinogen
TABLE 2 Proportion of Variance Explained by the Covariate With the Number of Subjects Within Parentheses

*p < .05; **p < .01.
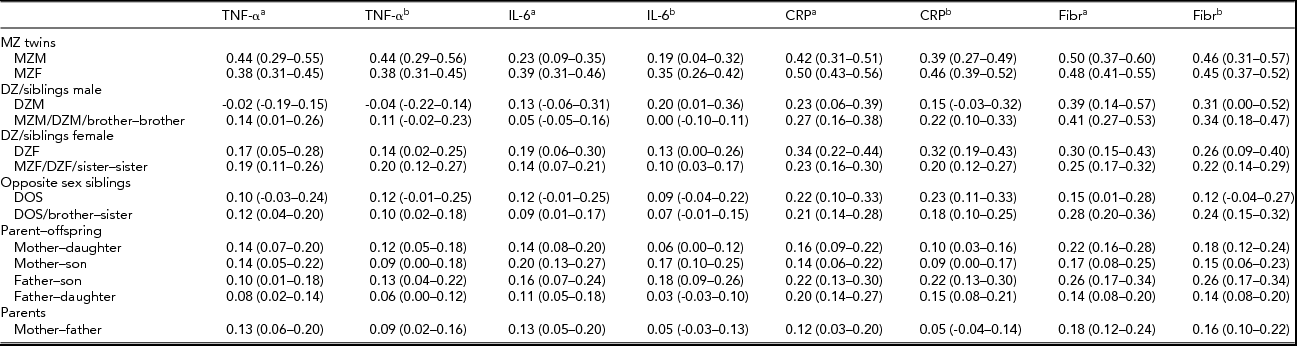
The effect of age on all parameters was significant and positive (p < .01). Sex differences were present only for CRP and fibrinogen with women having significantly larger mean values than men (p < .01). For TNF-α and CRP values, standard deviations were significantly larger in females than in males. Table 3 shows family correlations for each of the immune parameters estimated for the values adjusted only for age and sex and for the values additionally adjusted for other covariates.
TABLE 3 Family Correlations (and 95% Confidence Intervals) as Estimated in the Saturated Model for the Cytokines and the Acute Phase Reactants, With the Levels of the Pro-Inflammatory Markers Only Adjusted for Age and Sex, and Adjusted for All Covariates.
aAfter adjustment for the effects of age and sex; bafter adjustment for the effects of all covariates (age, sex, month, BMI, smoking, and for TNF-α and IL-6 plate as well).
For all parameters, male and female MZ correlations did not differ significantly and the same-sex and opposite-sex DZ twin and non-twin sibling correlations were also similar in all cases (p > .01), so no quantitative and qualitative sex differences were present, nor did we find evidence for twin-specific environmental effects. Parent–offspring correlations were not significantly different from DZ twin and non-twin sibling correlations (p > .01), except for the fibrinogen values adjusted for age and sex only (p = .003). This effect was not present in the fully adjusted fibrinogen values. These results suggest that genetic regulation of cytokine and acute phase reactant levels does not change significantly across age. Overall, adjustment for BMI, smoking, month, and plate effects, in addition to age and sex, tended to reduce all familial correlations, including the spouse correlations. Only the spouse correlation for the fully adjusted fibrinogen values was significantly different from zero (r = .16, p < .01), which indicates that the effects of assortative mating or sharing a household without being biologically related are negligible, except for the small resemblance found for fibrinogen.
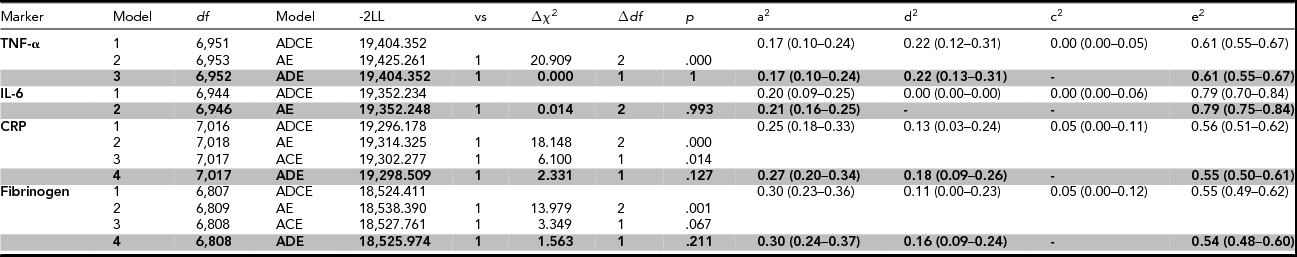
For the heritability analyses on the fully adjusted values, contributions of A, D, C, and E factors to the total phenotypic variance were constrained over sex while taking into account sex differences in phenotypic variance in TNF-α, CRP, and IL-6. Assortative mating was only modeled for fibrinogen. Table 4 shows the genetic models that were fitted to the data, supplemented with the proportions of the phenotypic variance that can be explained by different genetic and environmental factors for both the full ADCE model and the model that provided the most parsimonious fit.
TABLE 4 Genetic Model Fit Statistics of the Pro-Inflammatory Markers After Adjustment for Sex, Age, BMI, Smoking, Plate, and Month of Sampling
Abbreviations: df = degrees of freedom; Model = specification of the model that is tested; -2LL = minus twice the logarithm of the likelihood; vs = the model against which this submodel is tested; Δχ2 = model fit statistic: difference in -2LL of two nested models; ∆df = difference in the number of parameters between the models, p = p-value (was regarded significant when <.01), a2, d2, c2, e2 = proportions of variance explained by additive, non-additive, shared, and unique environmental effects. The 95% confidence intervals are depicted within parentheses. The most parsimonious model is printed in bold.
The broad-sense heritability was 39%, 21%, 45%, and 46% for TNF-α, Il-6, CRP, and fibrinogen respectively. The models that provided the best fit to the data on TNF-α, CRP, and fibrinogen included additive and non-additive genetic factors, and unique environmental factors. Non-additive genetic effects explained 22% of the variance of TNF-α, 18% of the variance of CRP, and 16% of the variance of fibrinogen. For CRP and fibrinogen, a small amount of variation was attributed to sibling-shared environmental factors in the full ADCE model, but an ADE model without shared environmental factors did not fit significantly worse. For IL-6, a model with additive genetic factors and unique environmental factors explained the data best, with no role for non-additive genetic factors, nor for shared environmental factors.
Discussion
This is the most comprehensive twin-family study of the genetic architecture of the pro-inflammatory state that has been performed thus far. Results replicate the importance of genetic factors in pro-inflammation observed before (de Lange et al., Reference de Lange, Snieder, Ariens, Spector and Grant2001, Reference de Lange, de Geus, Kluft, Meijer, van Doornen, Boomsma and Snieder2006; de Maat et al., Reference de Maat, Bladbjerg, Hjelmborg, Bathum, Jespersen and Christensen2004; Grunnet et al., Reference Grunnet, Poulsen, Klarlund, Mandrup-Poulsen and Vaag2006; Jermendy et al., Reference Jermendy, Horvath, Littvay, Steinbach, Jermendy, Tarnoki and Osztovits2011; MacGregor et al., Reference MacGregor, Gallimore, Spector and Pepys2004; Rahman et al., Reference Rahman, Bennet, Pedersen, de, Svensson and Magnusson2009; Reed et al., Reference Reed, Tracy and Fabsitz1994; Sas et al., Reference Sas, Jamshidi, Zheng, Wu, Korf, Alizadeh and Snieder2012; Su et al., Reference Su, Snieder, Miller, Ritchie, Bremner, Goldberg and Vaccarino2008, Reference Su, Lampert, Zhao, Bremner, Miller, Snieder and Vaccarino2009a, Reference Su, Miller, Snieder, Bremner, Ritchie, Maisano and Vaccarino2009b; Wang et al., Reference Wang, Christoffel, Brickman, Hong, Arguelles, Zhang and Wang2011; Wessel et al., Reference Wessel, Moratorio, Rao, Mahata, Zhang, Greene and O'Connor2007; Worns et al., Reference Worns, Victor, Galle and Hohler2006) and extend the findings of previous studies by showing that genetic non-additivity is an important factor in explaining individual differences in TNF-α, CRP, and fibrinogen levels and by ruling out a large role for environmental factors shared by family members.
There have been only three previous heritability studies employing a sample size of over 1,000 twins for CRP (Rahman et al., Reference Rahman, Bennet, Pedersen, de, Svensson and Magnusson2009; Wang et al., Reference Wang, Christoffel, Brickman, Hong, Arguelles, Zhang and Wang2011), IL-6 and TNF-α (Sas et al., Reference Sas, Jamshidi, Zheng, Wu, Korf, Alizadeh and Snieder2012). For fibrinogen, the study with the largest sample size included 962 subjects (de Lange et al., Reference de Lange, Snieder, Ariens, Spector and Grant2001). None of these studies systematically corrected for recent illness, medication use, menstrual cycle, oral contraceptives use, batch effects, month of sampling, BMI, and smoking status as done in the present study. In spite of the more strict correction for confounders, our heritability estimate for CRP was of comparable magnitude to these previous studies. For CRP, we confirmed the importance of non-additive genetic factors that was found in the larger of the two previous twin studies (Rahman et al., Reference Rahman, Bennet, Pedersen, de, Svensson and Magnusson2009), whereas the smaller of the two studies (Wang et al., Reference Wang, Christoffel, Brickman, Hong, Arguelles, Zhang and Wang2011) only detected additive effects, likely reflecting insufficient power. For fibrinogen, our broad-sense heritability estimate was comparable to the estimate reported by de Lange and colleagues (Reference de Lange, Snieder, Ariens, Spector and Grant2001), but our study additionally indicated that a significant proportion of the heritability was due to non-additive genetic effects.
For TNF-α and IL-6, our results do not completely support the results of the only large (N > 1,000) previous twin study (Sas et al., Reference Sas, Jamshidi, Zheng, Wu, Korf, Alizadeh and Snieder2012). For both cytokines, Sas and colleagues (Reference Sas, Jamshidi, Zheng, Wu, Korf, Alizadeh and Snieder2012) found substantial family resemblance, but they could not discriminate between genetics and shared environment as the source of that resemblance. The twin correlations reported in a smaller study on IL-6 (Worns et al., Reference Worns, Victor, Galle and Hohler2006) were suggestive of genetic factors and a potential role of shared environment with the MZ correlation being less than twice as high as the DZ correlation. Our study clearly shows that shared genetic make-up rather than shared family environment is the major source of familial resemblance in these parameters. Furthermore, we show a significant effect of non-additive factors on TNF-α. Four smaller twin studies of TNF-α and IL-6 values in healthy unchallenged subjects are consistent with our findings, as the MZ correlations found in those studies were about twice as high as the DZ correlations in elderly subjects (de Maat et al., Reference de Maat, Bladbjerg, Hjelmborg, Bathum, Jespersen and Christensen2004), young adults (Grunnet et al., Reference Grunnet, Poulsen, Klarlund, Mandrup-Poulsen and Vaag2006), and middle-aged twins (Su et al., Reference Su, Snieder, Miller, Ritchie, Bremner, Goldberg and Vaccarino2008, Reference Su, Miller, Snieder, Bremner, Ritchie, Maisano and Vaccarino2009b). In their sample of young adult subjects, Grunnet and colleagues (Reference Grunnet, Poulsen, Klarlund, Mandrup-Poulsen and Vaag2006) even found the MZ correlations for TNF-α to be more than twice as high as the DZ correlations.
Taken together, our results and those from previous studies suggest that about a third of the variation in these core pro-inflammatory cytokines and acute phase reactants in healthy subjects with values in the non-extreme range is explained by genetic variation. This means that some individuals are more susceptible than others to having higher levels of pro-inflammatory markers and this increased susceptibility is, at least partly, due to genetic differences between individuals. Large-scale collaborative attempts to find actual genes that underlie this genetic variation are underway. In 2011, a meta-analysis of genome-wide association (GWA) studies of CRP in over 80,000 subjects identified several genes implicated in the functioning and inflammation of immune system (CRP, IL6R, NLRP3, IL1F10, IRF1, PPP1R3B, SALL1, PABPC4, ASCL1, RORA, and BCL7B) and the metabolic syndrome (APOC1, HNF1A, LEPR, GCKR, HNF4A, and PTPN2) to be associated with circulating CRP levels (Dehghan et al., Reference Dehghan, Dupuis, Barbalic, Bis, Eiriksdottir, Lu and Chasman2011). In a meta-analysis of six GWA studies on fibrinogen in over 22,000 subjects, significant genome-wide hits were found in the FGB, IRF1, PCCB, and NLRP3 genes (Dehghan et al., Reference Dehghan, Yang, Peters, Basu, Bis, Rudnicka and Folsom2009). For IL-6 and TNF-α no meta-analysis has been published to our knowledge. A single large GWA study on IL-6 (N = 6,145) found significant hits in IL6R and ABO genes (Naitza et al., Reference Naitza, Porcu, Steri, Taub, Mulas, Xiao and Cucca2012). With our study, we have accomplished a clear numerical target for these ongoing genome-wide screens that aim to find the actual genetic variants regulating the levels of these pro-inflammatory markers. Heritability studies conducted in large representative samples continue to be valuable because the heritability of traits can vary between populations and can change across generations. We should also keep in mind that 54% to 79% of the variation found was due to unique environmental factors that are not shared between family members. This estimate may derive from unique environmental factors or measurement error, but it may also result from gene-by-environment interactions that may inflate estimates of unique environmental effects. Unravelling the genetics of these pro-inflammatory parameters may greatly contribute to our understanding of the etiology of cardiac disease and major depression as chronic low-grade inflammation has repeatedly been shown to be associated with both (Cesari et al., Reference Cesari, Penninx, Newman, Kritchevsky, Nicklas, Sutton-Tyrrell and Pahor2003; Danesh et al., Reference Danesh, Wheeler, Hirschfield, Eda, Eiriksdottir, Rumley and Gudnason2004, Reference Danesh, Kaptoge, Mann, Sarwar, Wood, Angleman and Gudnason2008; Humphries et al., Reference Humphries, Cooper, Talmud and Miller2007; Packard & Libby, Reference Packard and Libby2008; Libby & Theroux, Reference Libby and Theroux2005; O'Brien et al., Reference O'Brien, Scott and Dinan2004; Woods et al., Reference Woods, Brull, Humphries and Montgomery2000; Penninx et al., Reference Penninx, Kritchevsky, Yaffe, Newman, Simonsick, Rubin and Pahor2003).
Because of the large sample size and the extended twin-family design, this study had sufficient power to decompose the variance in the levels of an important set of pro-inflammatory markers into additive and non-additive genetic factors, and shared and unique environmental factors. The inclusion of parents and siblings allowed us to detect and correct for assortative mating, quantitative and qualitative sex differences, and effects of age that could potentially affect the heritability estimates. The sample size also allowed us to employ strict exclusion criteria concerning the recent health status and medication use of the subjects without losing power so that we were ensured that analyses were run on healthy individuals only. Furthermore, our study design controlled for female sex hormone status.
This study also had limitations. First, we selected only a subset of the many immune parameters that co-determine the pro-inflammatory state, including IL-1 and interferon-γ, and we did not take into account the action of soluble receptors for the cytokines, levels of which may be substantially heritable. Second, we used the ELISA by R&D systems that may yield higher TNF-α and IL-6 values in individuals with blood group O than other ABO blood groups, which may, in part, be due to assay-specific cross-reactivity with ABO antigens (Melzer et al., Reference Melzer, Perry, Hernandez, Corsi, Stevens, Rafferty and Ferrucci2008; Naitza et al., Reference Naitza, Porcu, Steri, Taub, Mulas, Xiao and Cucca2012). We indeed found a significant association of SNPs in the ABO region to TNF-α and IL-6. Although it explained only a small amount of variance in TNF-α (7%) and IL-6 (4%) compared with the larger effects of plate and BMI, they may cause overestimation of non-additivity or underestimation of shared environment as the shared blood group O will make MZ twins appear more alike than DZ twins or non-twin siblings. Third, we tested whether different genes are expressed at different ages by testing whether parent–offspring correlations and correlations in first-degree relatives (DZ twins and non-twin siblings) were of comparable magnitude. Because there was a partial overlap in age between the parent and the offspring generation, we cannot completely rule out the possibility that the expression of pro-inflammatory genes changes across age.
In conclusion, the familial resemblance in these core pro-inflammatory cytokines and acute phase reactants is explained by genetic variation and not by the shared family environment. For three out of four markers, both additive and non-additive genetic factors contribute to heritability.
Acknowledgments
We thank all twins and their family members for their participation in the Netherlands Twin Register Biobank project, and we also thank J. Neuteboom for technical assistance with the laboratory analyses. Funding was obtained from the Netherlands Organization for Scientific Research (NWO): Genotype/phenotype database for behavior genetic and genetic epidemiological studies (ZonMw Middelgroot 911–09–032) and by the European Science Council (ERC Advanced Grant 230374).