



Chordoma is a rare neoplasm derived from remnants of the fetal notochord, with incidence of approximately 1/1,000,000. Classic locations for chordoma include the lumbosacral spine (30-50%) and clivus (30-35%).Reference Erdem, Angtuaco and Van Hemert 1 Clival chordoma has a high rate of recurrence due to its aggressive biology as well as technical difficulty with gross total resection. These tumors frequently present with neuro-ophthalmologic findings.Reference Michele and Samuel 2 Intradural and extradural extension of clival chordoma are known phenomena, but brainstem involvement is exceedingly rare.
A 62-year-old man presenting with right-sided cranial nerve VI palsy was found on MRI to have a large sellar mass with invasion of the retroclival space, thought to be an aggressive pituitary macroadenoma. The patient was booked for endoscopic transsphenoidal resection of this tumor. Intraoperatively, the tumor was noted to erode through the floor of the sella turcica and involved bony destruction of the clivus, consistent with chordoma. The tumor was noted to extend through a small dural defect into the prepontine cistern. Near gross total resection of the tumor was achieved, except a small portion anterior to the brainstem. Final pathology revealed chordoma. The patient had two repeat transsphenoidal resections 10 months and 18 months after his initial debulking, with intraoperative note made of brainstem invasion. He also received radiation therapy following his third surgery. He was then followed with MRI to monitor recurrence at 6-month intervals.
The patient remained free of recurrence until 1 year after his third operation. At that time radiologic recurrence was noted with only minor symptoms. After 6 months, the patient developed lower cranial nerve deficits including swallowing issues and drop attacks. An attempt at surgical decompression was undertaken via a retrosigmoidal approach, but the tumor had become too adherent to the brainstem, extending from the prepontine cistern into the belly of the pons with cystic component (Figure 1), with adjacent signal changes on FLAIR and T2 images. Diffusion weighted imaging showed some areas of restriction suggestive of infarction. The patient passed away during admission, likely from brainstem perforator infarct (Figures 2-4).

Figure 1 Coronal and sagittal post-gadolinium T1 magnetic resonance images showing clival chordoma at the time of diagnosis. The tumor demonstrates destruction of the clivus and involvement of the prepontine cistern but no intrapontine extension.
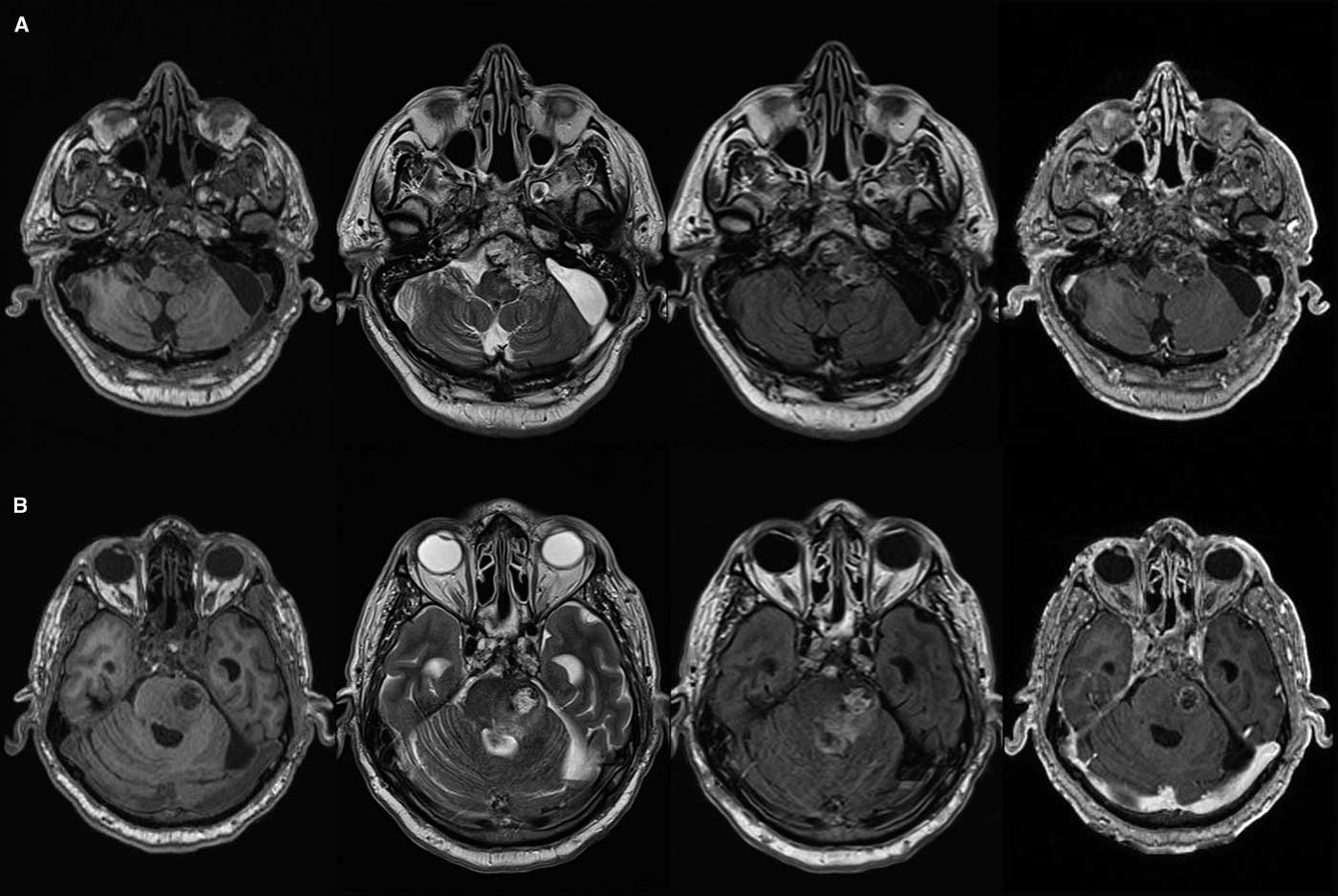
Figure 2 Axial T1, T2, FLAIR and post-gadolinium T1 magnetic resonance images showing clival chordoma and T2/FLAIR signal change in prepontine cistern (A) with intrapontine invasion (B).
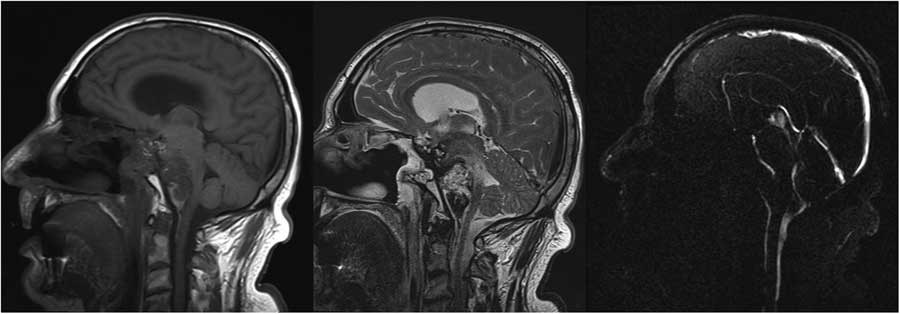
Figure 3 Sagittal T1, T2 and PC in plane flow magnetic resonance images demonstrating pontine infiltration of the tumor and compression of the basilar artery.

Figure 4 Axial T2, FLAIR, diffusion weighted imaging and ADC images showing areas of restricted diffusion within the brainstem.
Brainstem invasion of clival chordoma is exceedingly rare, with only four cases reported in the literature.Reference Herold, Giordano, Naka, Gerganov, Samii and Samii 3 - Reference Zoli, Milanese and Bonfatti 5 Aggressive extradural and intradural involvement is well known. Occasionally, chordoma can occur as a primary intradural lesion, which can be quite benign with good outcomes, though can rarely exhibit aggressive character.Reference Ito, Saito and Nagatani 6 Other differential diagnosis for chordoma includes fetal notochord, ecchordosis physaliphora, atypical meningioma and chondrosarcoma.Reference Erdem, Angtuaco and Van Hemert 1
Treatment of chordoma is by surgical debulking. Conventional radiotherapy adds no benefit to treatment of these tumors, though postoperative proton therapy has been shown to improve survival.Reference Colli and Al-Mefty 7 Surgical approaches include transsphenoidal, transcranial, transoropharygeal and maxillary osteotomy.Reference Colli and Al-Mefty 7 The endoscopic transsphenoidal approach represents a useful, minimally invasive option with good visualization of the sella, clivus and retroclivus.Reference Ito, Saito and Nagatani 6 , Reference Hong Jiang, Ping Zhao, Hai Xie, Zhang, Zhang and Yun Xiao 8 , Reference Carrabba, Dehdashti and Gentili 9 Patients with chordoma require extensive follow-up due to high risk for recurrence.
Clival chordoma is a rare intracranial neoplasm with many presentations. Extensive follow-up is required for patients with these tumors. Posterior fossa involvement and especially brainstem extension are associated with several catastrophic complications, including basilar artery compression and lower cranial nerve deficits.
Disclosure
Amit R. L. Persad, Bradford Mechor and Yves Starreveld have no disclosures.




