



Introduction
The common hallmark of almost all cancers and tumours is rapid, uncontrolled growth, cellular proliferation and metastasis (Yamamoto et al., Reference Yamamoto, Seino, Fukumoto, Koh, Yano, Inagaki, Yamada, Inoue, Manabe and Imura1990; Macheda et al., Reference Macheda, Rogers and Best2005; Airley and Mobasheri, Reference Airley and Mobasheri2007; Szablewski, Reference Szablewski2013; Wang et al., Reference Wang, Xiao, Li, Aziz, Gan, Johnson and Chen2015; Barron et al., Reference Barron, Bilan, Tsakiridis and Tsiani2016; Ancey et al., Reference Ancey, Contat and Meylan2018, Wu et al., Reference Wu, Xu, Liang, Meng, Hua, Wang, Zhang, Liu, Yu and Shi2021). Such metastatic growth demands a constant supply of nutrients, especially sugars, particularly glucose and fructose and most cancer cells upregulate their glucose transporters (GLUTs) without obvious mutations (Godoy et al., Reference Godoy, Ulloa, Rodríguez, Reinicke, Yañez, García Mde, Medina, Carrasco, Barberis, Castro, Martínez, Koch, Vera, Poblete, Figueroa, Peruzzo, Pérez and Nualart2006; Barron et al., Reference Barron, Bilan, Tsakiridis and Tsiani2016). It has been shown that glucose transporters GLUT1–14, especially GLUT 1–9, 11–12, 14 are involved in cancer metabolism and fuel cancer growth (Barron et al., Reference Barron, Bilan, Tsakiridis and Tsiani2016; Ancey et al., Reference Ancey, Contat and Meylan2018). If these sugar transporters can be effectively targeted and specifically inhibited, the proliferation and metastasis of cancer cells may also likely be inhibited. Structural studies of these GLUTs may ultimately lead to significant clinical benefits for cancer patients.
GLUTs have different effects and involvement in various cancers (Table 1; Chandler et al., Reference Chandler, Williams, Slavin, Best and Rogers2003; Barron et al., Reference Barron, Bilan, Tsakiridis and Tsiani2016). For example, overexpression or upregulation of GLUT1, GLUT3, GLUT5 and GLUT12 is widely found in many types of cancers including breast, bladder, cervical, colon, colorectal, oesophageal, glioblastoma, gastric, head and neck, laryngeal, liver, lung, lymphoma, oral squamous cell, ovarian, pancreas, pancreatic islets, penile, prostate, clear renal cell, testis, thyroid, uterine cancers as well as from breast cancer to brain metastasis (Nishioka et al., Reference Nishioka, Oda, Seino, Yamamoto, Inagaki, Yano, Imura, Shigemoto and Kikuchi1992; Brown and Wahl, Reference Brown and Wahl1993; Mellanen et al., Reference Mellanen, Minn, Grenman and Harkonen1994; Zamora-Leon et al., Reference Zamora-León, Golde, Concha, Rivas, Delgado-López, Baselga, Nualart and Vera1996; Higashi et al., Reference Higashi, Tamaki, Honda, Torizuka, Kimura, Inokuma, Ohshio, Hosotani, Imamura and Konishi1997; Younes et al., Reference Younes, Brown, Stephenson, Gondo and Cagle1997; Noguchi et al., Reference Noguchi, Marat, Saito, Yoshikawa, Doi, Fukuzawa, Tsuburaya, Satoh and Ito1999; Pedersen et al., Reference Pedersen, Holm, Lund, Højgaard and Kristjansen2001; Rogers et al., Reference Rogers, Docherty, Slavin, Henderson and Best2003; Ayala et al., Reference Ayala, Rocha, Carvalho, Carvalho, da Cunha, Lourenço and Soares2010; Medina Villaamil et al., Reference Medina Villaamil, Aparicio Gallego, Valbuena Rubira, García Campelo, Valladares-Ayerbes, Grande Pulido, Victoria Bolós, Santamarina Caínzos and Antón Aparicio2011; Starska et al., Reference Starska, Forma, Józwiak, Brys, Lewy-Trenda, Brzezinska-Blaszczyk and Krzeslak2015; Barron et al., Reference Barron, Bilan, Tsakiridis and Tsiani2016; Schlößer et al., Reference Schlößer, Drebber, Urbanski, Haase, Baltin, Berlth, Neiß, von Bergwelt-Baildon, Fetzner, Warnecke-Eberz, Bollschweiler, Hölscher, Mönig and Alakus2017; White et al., Reference White2018; Kuo et al., Reference Kuo, Chang, Yeh, Chu, Lee and Lee2019; Heydarzadeh et al., Reference Heydarzadeh, Moshtaghie, Daneshpoor and Hedayati2020; Achalandabaso-Boira et al., Reference Achalandabaso-Boira, Di Martino, Gordillo, Adrados and Martín-Pérez2020). Overexpression of GLUT2 is found in cancers of the breast, colon, liver, pancreas and small intestine (Tomita, Reference Tomita1999; Godoy et al., Reference Godoy, Ulloa, Rodríguez, Reinicke, Yañez, García Mde, Medina, Carrasco, Barberis, Castro, Martínez, Koch, Vera, Poblete, Figueroa, Peruzzo, Pérez and Nualart2006; Hamann et al., Reference Hamann, Krys, Glubrecht, Bouvet, Marshall, Vos, Mackey, Wuest and Wuest2018). Overexpression of GLUT4 is found in breast, gastric, and myeloma, muscle (McBrayer et al., Reference McBrayer, Cheng, Singhal, Krett, Rosen and Shanmugam2012; Guo et al. 2021); overexpression of GLUT6 is found in gastric and endometrial cancer and testis (Byrne et al., Reference Byrne, Poon, Modesitt, Tomsig, Chow, Healy, Baker, Atkins, Lancaster, Marchion, Moley, Ravichandran, Slack-Davis and Hoehn2014; Schlößer et al., Reference Schlößer, Drebber, Urbanski, Haase, Baltin, Berlth, Neiß, von Bergwelt-Baildon, Fetzner, Warnecke-Eberz, Bollschweiler, Hölscher, Mönig and Alakus2017; Caruana and Byrne, Reference Caruana and Byrne2020); overexpression of GLUT7 is found in benign prostate cancer (Reinicke et al., Reference Reinicke, Sotomayor, Cisterna, Delgado, Nualart and Godoy2012); overexpression of GLUT8 is found in myeloma (McBrayer et al., Reference McBrayer, Cheng, Singhal, Krett, Rosen and Shanmugam2012); overexpression of GLUT9 is found in adrenal, heart, kidney, liver, both benign and cancerous prostate (Godoy et al., Reference Godoy, Ulloa, Rodríguez, Reinicke, Yañez, García Mde, Medina, Carrasco, Barberis, Castro, Martínez, Koch, Vera, Poblete, Figueroa, Peruzzo, Pérez and Nualart2006); overexpression of GLUT11 is found in multiple myeloma and prostate (McBrayer et al., Reference McBrayer, Cheng, Singhal, Krett, Rosen and Shanmugam2012) and overexpression of GLUT14 is found in colon and glioblastoma (Valli et al., Reference Valli, Morotti, Zois, Albers, Soga, Feldinger, Fischer, Frejno, McIntyre, Bridges, Haider, Buffa, Baban, Rodriguez, Yanes, Whittington, Lake, Zervou, Lygate, Kessler and Harris2019; Sharpe et al., Reference Sharpe, Ijare, Baskin, Baskin, Baskin and Pichumani2021). On the other hand, overexpression of GLUT10, GLUT12 and GLUT13 have also been found to be associated with better outcomes in lung adenocarcinoma (Du et al., Reference Du, Liu, Yuan, Zhang and Geng2020). Furthermore, a recent study showed that GLUT12 is expressed in insulin-sensitive tissues adipose tissue and it perhaps could modulate sugar absorption in physiological and pathophysiological obesity (Gil-Iturbe et al., Reference Gil-Iturbe, Arbones-Mainar, Moreno-Aliaga and Lostao2019).
Table 1. The 14 glucose transporter gene expressions in various cancers
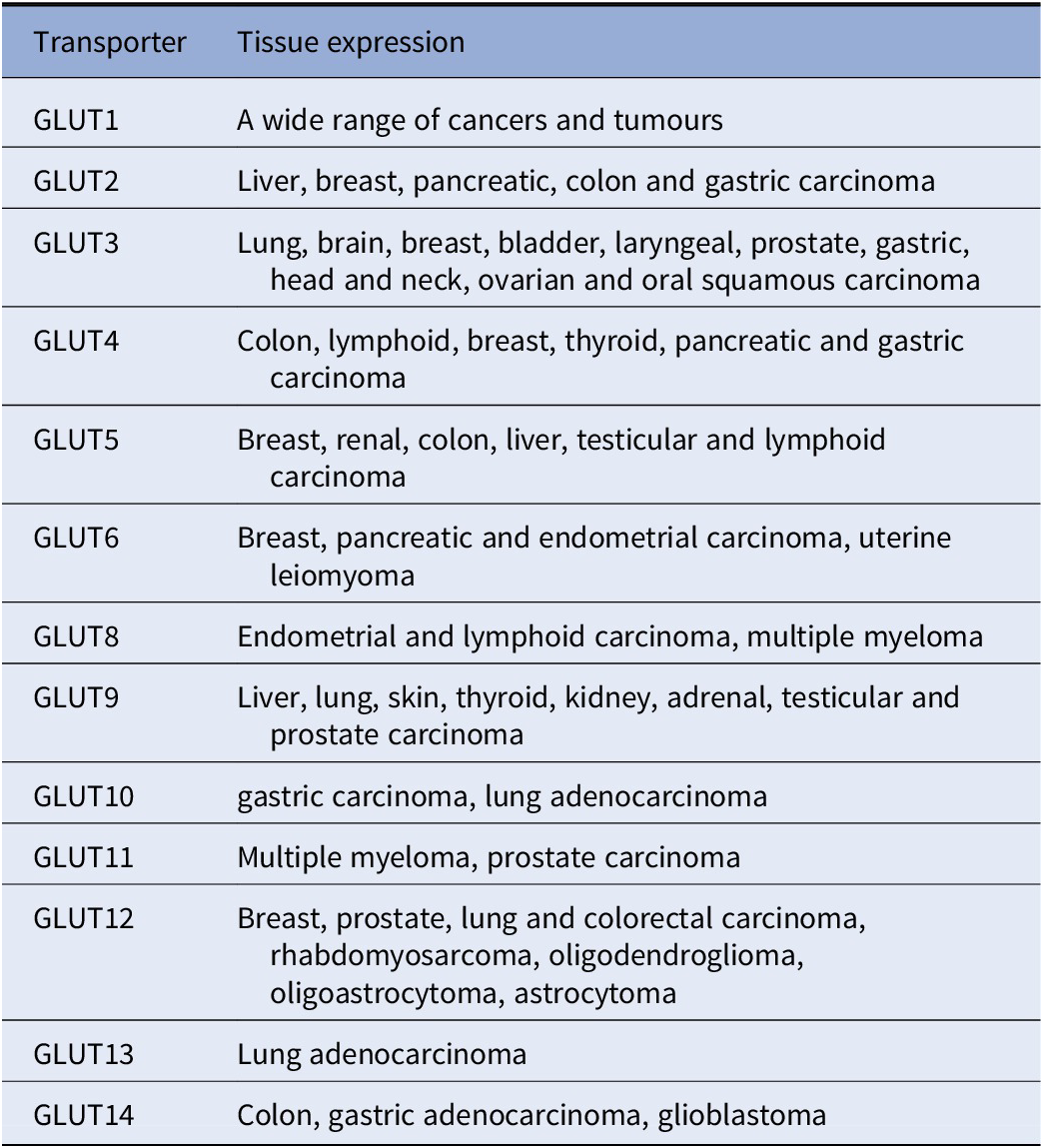
Note: This table is mainly derived from Barron et al. (Reference Barron, Bilan, Tsakiridis and Tsiani2016) and Wu et al. (Reference Wu, Xu, Liang, Meng, Hua, Wang, Zhang, Liu, Yu and Shi2021), except information for GLUT10 (Schlößer et al., Reference Schlößer, Drebber, Urbanski, Haase, Baltin, Berlth, Neiß, von Bergwelt-Baildon, Fetzner, Warnecke-Eberz, Bollschweiler, Hölscher, Mönig and Alakus2017; Du et al., Reference Du, Liu, Yuan, Zhang and Geng2020). GLUT 13 (Du et al., Reference Du, Liu, Yuan, Zhang and Geng2020) and GLUT14 (Berlth et al., Reference Berlth, Mönig, Pinther, Grimminger, Maus, Schlösser, Plum, Warnecke-Eberz, Harismendy, Drebber, Bollschweiler, Hölscher and Alakus2015; Valli et al., Reference Valli, Morotti, Zois, Albers, Soga, Feldinger, Fischer, Frejno, McIntyre, Bridges, Haider, Buffa, Baban, Rodriguez, Yanes, Whittington, Lake, Zervou, Lygate, Kessler and Harris2019; Sharpe et al., Reference Sharpe, Ijare, Baskin, Baskin, Baskin and Pichumani2021).
Thus far, only the molecular structures of GLUT1, GLUT3 and GLUT4 have been elucidated (Deng et al., Reference Deng, Sun, Yan, Ke, Jiang, Xiong, Ren, Hirata, Yamamoto, Fan and Yan2015; Custódio et al., Reference Custódio, Paulsen, Frain and Pedersen2021; Yuan et al., Reference Yuan, Kong, Xu, Zhu, Yan and Yan2022). The molecular structures of other twelve GLUTs remain to be determined. Since these transporters form 12 transmembrane helices, the structural determination of these transporters requires systematic detergent screens before protein purification can be carried out. The difficulties in obtaining structures of transmembrane species experimentally are well-known (Vinothkumar and Henderson, Reference Vinothkumar and Henderson2010).
AlphaFold2 and RoseTTAFold were introduced in July 2021 as an artificial intelligence (AI) revolutionary computational tool for the accurate prediction of protein structures (Baek et al., Reference Baek, DiMaio, Anishchenko, Dauparas, Ovchinnikov, Lee, Wang, Cong, Kinch, Schaeffer, Millán, Park, Adams, Glassman, DeGiovanni, Pereira, Rodrigues, van Dijk, Ebrecht, Opperman, Sagmeister, Buhlheller, Pavkov-Keller, Rathinaswamy, Dalwadi, Yip, Burke, Garcia, Grishin, Adams, Read and Baker2021; Jumper et al., Reference Jumper, Evans, Pritzel, Green, Figurnov, Ronneberger, Tunyasuvunakool, Bates, Žídek, Potapenko, Bridgland, Meyer, Kohl, Ballard, Cowie, Romera-Paredes, Nikolov, Jain, Adler, Back, Petersen, Reiman, Clancy, Zielinski, Steinegger, Pacholska, Berghammer, Bodenstein, Silver, Vinyals, Senior, Kavukcuoglu, Kohli and Hassabis2021; Varadi et al., Reference Varadi, Anyango, Deshpande, Nair, Natassia, Yordanova, Yuan, Stroe, Wood, Laydon, Žídek, Green, Tunyasuvunakool, Petersen, Jumper, Clancy, Green, Vora, Lutfi, Figurnov, Cowie, Hobbs, Kohli, Kleywegt, Birney, Hassabis and Velankar2022). Since its introduction, both AlphaFold2 and RoseTTAFold have already made a significant impact on our understanding of the molecular structure of numerous proteins that were previously inaccessible. However, investigators, biotech, and the pharmaceutical industry are still very interested in studying the physical structures of proteins, especially membrane transporters, since the structures are vital to understanding how glucose and other sugars are transported across cell membranes.
We previously applied the glutamine, threonine, tyrosine (QTY) code to design several detergent-free transmembrane (TM) protein chemokine receptors and cytokine receptors for various uses using conventional computing programs (Zhang et al., Reference Zhang, Tao, Qing, Tang, Skuhersky, Corin, Tegler, Wassie, Wassie, Kwon, Suter, Entzian, Schubert, Yang, Labahn, Kubicek and Maertens2018). The expressed proteins exhibited predicted characteristics and retained ligand-binding activity (Zhang et al., Reference Zhang, Tao, Qing, Tang, Skuhersky, Corin, Tegler, Wassie, Wassie, Kwon, Suter, Entzian, Schubert, Yang, Labahn, Kubicek and Maertens2018; Qing et al., Reference Qing, Han, Skuhersky, Chung, Badr, Schubert and Zhang2019, Reference Qing, Tao, Chatterjee, Yang, Han, Chung, Ni, Suter, Kubicek, Maertens, Schubert, Blackburn and Zhang2020; Tao et al., Reference Tao, Tang, Zhang, Li and Xu2022; Hao et al., Reference Hao, Jin, Zhang and Qing2020; Tegler et al., Reference Tegler, Corin, Skuhersky, Pick, Vogel and Zhang2020). Later we prepared QTY variant protein structure predictions using AlphaFold2, achieving results in hours (Skuhersky et al., Reference Skuhersky, Tao, Qing, Smorodina, Jin and Zhang2021) rather than 4–5 weeks for each molecular simulation using GOMoDo, AMBER and YASARA programs (Zhang et al., Reference Zhang, Tao, Qing, Tang, Skuhersky, Corin, Tegler, Wassie, Wassie, Kwon, Suter, Entzian, Schubert, Yang, Labahn, Kubicek and Maertens2018; Qing et al., Reference Qing, Han, Skuhersky, Chung, Badr, Schubert and Zhang2019; Tegler et al., Reference Tegler, Corin, Skuhersky, Pick, Vogel and Zhang2020). Here, we use AlphaFold2 to design water-soluble QTY variants of the 14 GLUTs, and to make comparison with the native structure. In addition to targeting the glucose uptake activity of cancer cells, the motivation to design these water-soluble GLUTs is to find many additional applications, such as ultrasensitive glucose sensing devices, as water-soluble antigens to generate valuable and specific therapeutic monoclonal antibodies to block cancer cell energy supplies. Working with water-soluble QTY variants may substantially accelerate the discovery and development of therapeutic and diagnostic biologicals.
Materials and methods
Protein sequence alignments and other characteristics
The native sequences for GLUT1 through GLUT14 and their QTY-variant sequences were aligned using the same methods previously described (Zhang et al., Reference Zhang, Tao, Qing, Tang, Skuhersky, Corin, Tegler, Wassie, Wassie, Kwon, Suter, Entzian, Schubert, Yang, Labahn, Kubicek and Maertens2018; Qing et al., Reference Qing, Han, Skuhersky, Chung, Badr, Schubert and Zhang2019). The molecular weights (MW) and pI values of the proteins were calculated using the service provided by Expasy (https://web.expasy.org/compute_pi/). For detailed transporter protein sequence information, the predicted 2D structures and hydrophobicity change, please see Supplementary Fig. 1.
AlphaFold2 predictions
Structure predictions of the QTY variants were performed using the AlphaFold2 (Jumper et al., Reference Jumper, Evans, Pritzel, Green, Figurnov, Ronneberger, Tunyasuvunakool, Bates, Žídek, Potapenko, Bridgland, Meyer, Kohl, Ballard, Cowie, Romera-Paredes, Nikolov, Jain, Adler, Back, Petersen, Reiman, Clancy, Zielinski, Steinegger, Pacholska, Berghammer, Bodenstein, Silver, Vinyals, Senior, Kavukcuoglu, Kohli and Hassabis2021; Varadi et al., Reference Varadi, Anyango, Deshpande, Nair, Natassia, Yordanova, Yuan, Stroe, Wood, Laydon, Žídek, Green, Tunyasuvunakool, Petersen, Jumper, Clancy, Green, Vora, Lutfi, Figurnov, Cowie, Hobbs, Kohli, Kleywegt, Birney, Hassabis and Velankar2022) software following the instructions at the website https://github.com/sokrypton/ColabFold on 2 × 20 Intel Xeon Gold 6248 cores, 384 GB RAM, and a Nvidia Volta V100 GPU. Other AlphaFold2-predicted structures were obtained from the European Bioinformatics Institute (EBI, https://alphafold.ebi.ac.uk) and are also available at Uniprot website https://www.uniprot.org. Each UniProt ID from the dataset was extended with ID, entry name, description, and FASTA sequence. The data was taken from UniProt using a custom Python code. The FASTA sequences were converted into their soluble versions using the QTY method (https://pss.sjtu.edu.cn/), followed by Protter 2D diagram plotting (http://wlab.ethz.ch/protter/start/). These steps were optimised via Python libraries for web applications such as requests and splinter.
Superposed structures
The published X-ray crystal structures of native GLUT1 (PDB: 6THA , 2.4 Å) and GLUT3 (PDB: 4ZW9, 1.5 Å) were obtained from the protein data bank (PDB), https://www.rcsb.org. AlphaFold2 predictions of 12 native GLUTs and their QTY variants were carried out using the AlphaFold2 program at https://github.com/sokrypton/ColabFold. All 14 native GLUT sequences are obtained from Uniprot https://www.uniprot.org. The X-ray crystal structures and predicted structures were aligned with PyMOL.
Structure visualisation
We used two programs for structure visualisation: PyMOL https://pymol.org/2/ and UCSF Chimera https://www.rbvi.ucsf.edu/chimera/. All superposed models were produced via PyMOL, whilst Chimera was used for hydrophobicity representation.
Data availability of AlphaFold2 predicted water-soluble QTY variants
The AlphaFold2 predicted protein structures are at EBI (https://alphafold.ebi.ac.uk; Table 2). The QTY code designed water-soluble GLUT1–14 variants are reported in this paper and for more detail information, please go to the website: https://github.com/eva-smorodina/glucose-transporters.
Table 2. AlphaFold2 structure prediction parameters

Results and discussions
Protein sequence alignments and other characteristics
We aligned the native GLUTs with their QTY variants. Despite significant QTY replacement of hydrophobic residues overall (~17–26%), especially in the transmembrane domains (~44–50%) in the GLUTs, the isoelectric-focusing point pI and molecular weight remain rather similar (Figs 1 and 2 and Table 3). This is because Q, T, Y amino acids do not introduce any charges, they only introduce water-soluble side chains. Q (glutamine) side chains form four water hydrogen bonds, two donors through ▬NH2, and two acceptors through oxygen on ▬C〓O; the sidechains ▬OH of T (threonine) and Y (tyrosine) form three water hydrogen bonds, one donor from H (hydrogen) and two acceptors from O (oxygen).

Fig. 1. Protein sequences alignments of native GLUT1 and GLUT3 with water-soluble QTY variants. The Q, T and Y amino acid substitutions are in red. The alpha-helical segments (blue) are shown above the protein sequences, the internal (yellow) and external (red) loops of the transporters are indicated. The symbols | and * indicate the identical and different amino acids, respectively. Characteristics of natural and QTY variants with pI, molecular weight, total variation rate and transmembrane variation rate are presented (also see Table 3). The alignment: (a) GLUT1 and GLUT1QTY, (b) GLUT3 and GLUT3QTY. Although there are significant overall changes, >26% for GLUT1 and >24% for GLUT3, the transmembrane (TM) domain changes, >48% for GLUT1 and >44% for GLUT3, their pI and molecular weight changes are minimal.

Fig. 2. Protein sequences alignment of 12 native glucose transporters, GLUT2 and from GLUT4 to GLUT14, with their water-soluble QTY variants. The Q, T and Y amino acid substitutions are in red. The alpha-helical segments (blue) are shown above the protein sequences, the external (red) and internal (yellow) loops of the receptors are indicated. The symbols | and * indicate the similar and different amino acids, respectively. Characteristics of natural and QTY variants with pI, molecular weight, total variation rate and membrane variation rate are presented (also see Table 1). The alignment: (a) GLUT2 and GLUT2QTY, (b) GLUT4 and GLUT4QTY, (c) GLUT5 and GLUT5QTY, (d) GLUT6 and GLUT6QTY, (e) GLUT7 and GLUT7QTY, (f) GLUT8 and GLUT8QTY, (g) GLUT9 and GLUT9QTY, (h) GLUT10 and GLUT10QTY, (i) GLUT11 and GLUT11QTY, (j) GLUT12 and GLUT12QTY, (k) GLUT13 and GLUT13QTY and (l) GLUT14 and GLUT14QTY.
Table 3. Characteristics of native glucose transporters and their water-soluble QTY variants

Abbreviations: MW, molecular weight; pI, isoelectric focusing; RMSD, residue mean-square distance in Å; TM, transmembrane; —, not applicable.
The QTY code selects three neutrally polar amino acids: glutamine (Q), threonine (T) and tyrosine (Y) to replace four hydrophobic amino acids leucine (L), isoleucine (I), valine (V) and phenylalanine (F), since their electron density maps share remarkable structure similarities between L versus Q, I,V versus T and F versus Y (Zhang et al., Reference Zhang, Tao, Qing, Tang, Skuhersky, Corin, Tegler, Wassie, Wassie, Kwon, Suter, Entzian, Schubert, Yang, Labahn, Kubicek and Maertens2018). After applying the QTY code, the hydrophobic amino acids in the transmembrane segments are replaced by Q, T and Y, therefore the transmembrane segments are no longer hydrophobic. For example, the overall proteins, GLUT1 differs 26.83% from its QTY variant and GLUT13 differs 17.28% from its QTY variant. Most differences are >20% (Figs 1 and 2 and Table 3).
The isoelectric-focusing points (pI) vary, some in the acidic and some in the basic range. For example, native GLUT1, GLUT2, GLUT6, GLUT7, GLUT9, GLUT10, GLUT11, GLUT12 and GLUT14 have basic pIs > 8.0. On the other hand, GLUT5 and GLUT13 have acidic pIs < 6.0. Others including GLUT3, GLUT4, GLUT8 have near neutral pIs, ~ 6.84–7.5 (Table 3). Notably, the isoelectrical focusing points (pIs) are identical for the native and QTY variants for GLUT4, GLUT5 and GLUT13 despite significant QTY sequence replacement. The pIs in the QTY variants have little changes, and in some examples, there are no change. This is because that three amino acids Q, T, Y do not bear any charges at neutral pH. Thus, introductions of these amino acids Q, T, Y do not significantly alter the pI. This is important since altered pI may result the non-specific interactions.
Furthermore, although there are between ~43.6 and ~50% transmembrane QTY replacements, the molecular weights of the native and QTY variants differ by only a few hundreds of Daltons. This is because the addition of −OH groups from QTY amino acids increased their molecular weights (Figs 1 and 2 and Table 3).
Superposition of native transporters and their water-soluble QTY variants
In this study, we compare the molecular structures of native GLUTs and their QTY variants. Since the crystal structures of native receptors GLUT1 (PDB: 6THA ) (Deng et al., Reference Deng, Sun, Yan, Ke, Jiang, Xiong, Ren, Hirata, Yamamoto, Fan and Yan2015) and GLUT3 (PDB: 4ZW9 ) (Custódio et al., Reference Custódio, Paulsen, Frain and Pedersen2021) are already available, the superposed structures were carried out for: GLUT1 versus GLUT1QTY, and GLUT3 versus GLUT3QTY. Therefore, their crystal structures and the AlphaFold2 predicted QTY variant structures can be directly compared.
The native structures and water-soluble QTY variants superposed very well, the RMSD is GLUT1 and GLUT1QTY is 1.545 Å and for GLUT3 and GLUT3QTY is 1.025 Å (Fig. 3, Table 3 and Supplementary Table 1). In the first sets of structures for GLUT1 and GLUT3, they are superposed among: (i) the X-ray crystal determined structures (magenta) GLUT1 ( 6THA ) and GLUT3 ( 4ZW9 ), and the (ii) AlphaFold2 predicted water-soluble QTY variants (cyan). As seen from Fig. 3, these structures clearly superposed well, the structures are viewed from front, back, top and bottom. The results suggest that these structures share very similar folds despite >44% QTY amino acid replacement in the transmembrane helices in the water-soluble QTY variants. These closely superposed structures perhaps confirm that the AlphaFold2’s predictions are highly accurate, since the predicted native structures are directly superposable with the experimentally determined X-ray crystal structures. These results also suggest the native GLUT and their water-soluble QTY variants share remarkable structural similarity.
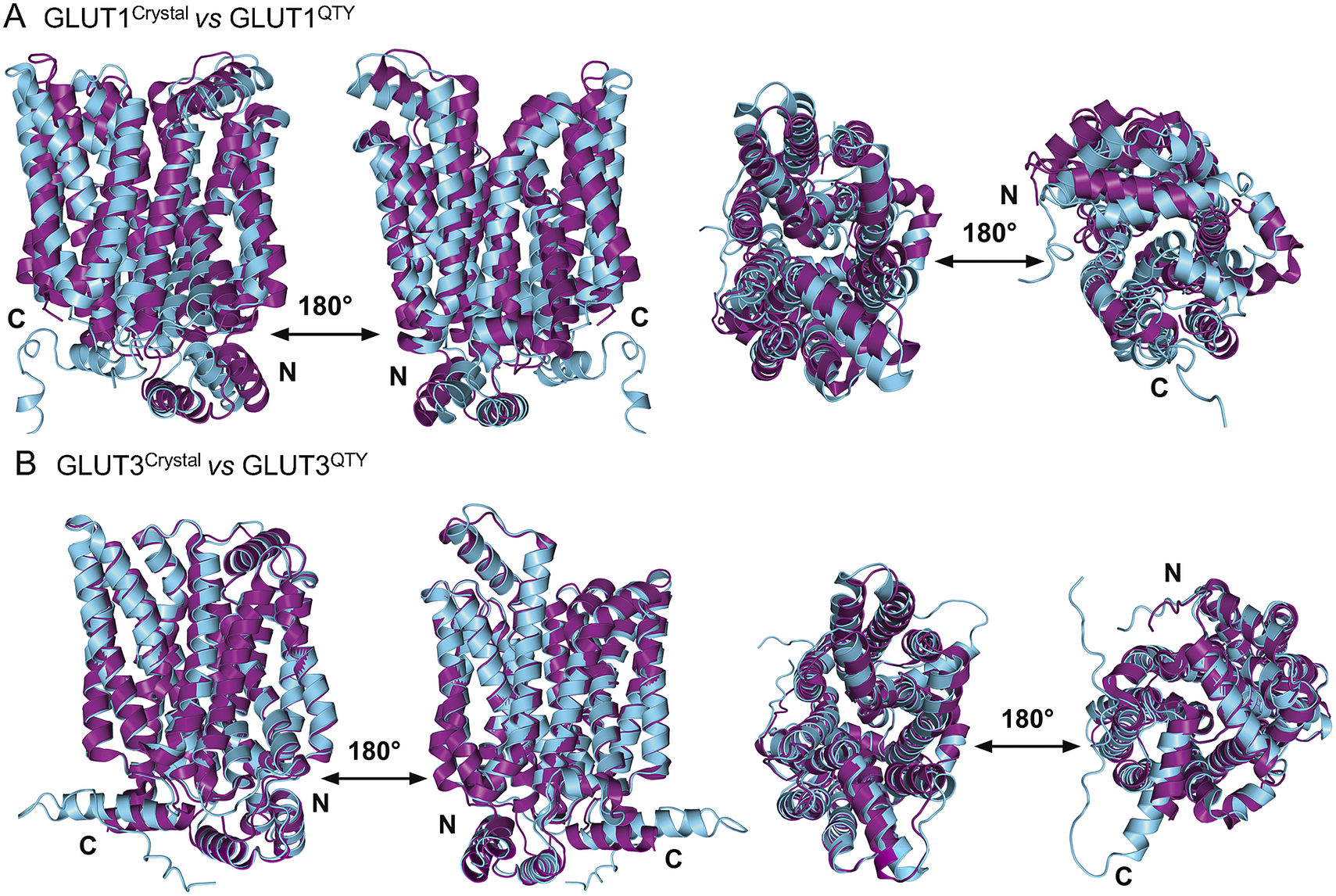
Fig. 3. Superposed two transporter crystal structures of GLUT1Crystal and GLUT3Crystal with AlphaFold2 predicted QTY water-soluble variants GLUT1QTY and GLUT3QTY. For each superposition, four structures are shown, front (left), back (middle left) and view from top axis (middle right) and bottom (right). The X-ray crystal structures of native GLUT1 (6THA, 2.4 Å, P11166), GLUT3 (4ZW9, 1.5 Å, P11169), are obtained from the protein data bank (PDB). N- and C-termini are labelled. (a) The crystal structure GLUT1Crystal (magenta) is superposed with AlphaFold2 predicted water-soluble variant GLUT1QTY (cyan). The RMSD is 1.545 Å for GLUT1 and GLUT1QTY. Following the same order, the superposed crystal structure and AlphaFold2 predicted structures of (b) GLUT3Crystal (magenta), water-soluble variant GLUT3QTY (cyan). The RMSD is 1.025 Å for GLUT3Crystal and GLUT3QTY. These results show that the glucose transporter water-soluble QTY variants share remarkable structural similarity despite >44% QTY replacements in transmembrane alpha-helices.
Since the molecular structures for the other 12 native glucose transporters (GLUT2, and GLUT4 to GLUT14) are not yet available, we used AlphaFold2 predictions. The residue mean-square distances (RMSD in Å) for the superposed structures are also shown in Fig. 4 and Table 3. The pairwise examples are: GLUT1 versus GLUT1QTY (1.545 Å), GLUT2 versus GLUT2QTY (3.058 Å), GLUT3 versus GLUT3QTY (1.025 Å), GLUT4 versus GLUT4QTY (0.764 Å), GLUT5 versus GLUT5QTY (0.712 Å), GLUT6 versus GLUT6QTY (1.910 Å), GLUT7 versus GLUT7QTY (0.470 Å), GLUT8 versus GLUT8QTY (1.169 Å), GLUT9 versus GLUT9QTY (0.593 Å), GLUT10 versus GLUT10QTY (1.186 Å), GLUT11 versus GLUT11QTY (1.502 Å), GLUT12 versus GLUT12QTY (3.590 Å), GLUT13 versus GLUT13QTY (0.881 Å) and GLUT14 versus GLUT14QTY (2.720 Å) (Table 3 and Supplementary Table 2). These AlphaFold2-predicted native and their water-soluble GLUT structures superposed very well suggesting that they share similar molecular structures despite the large transmembrane variations (44–50%).
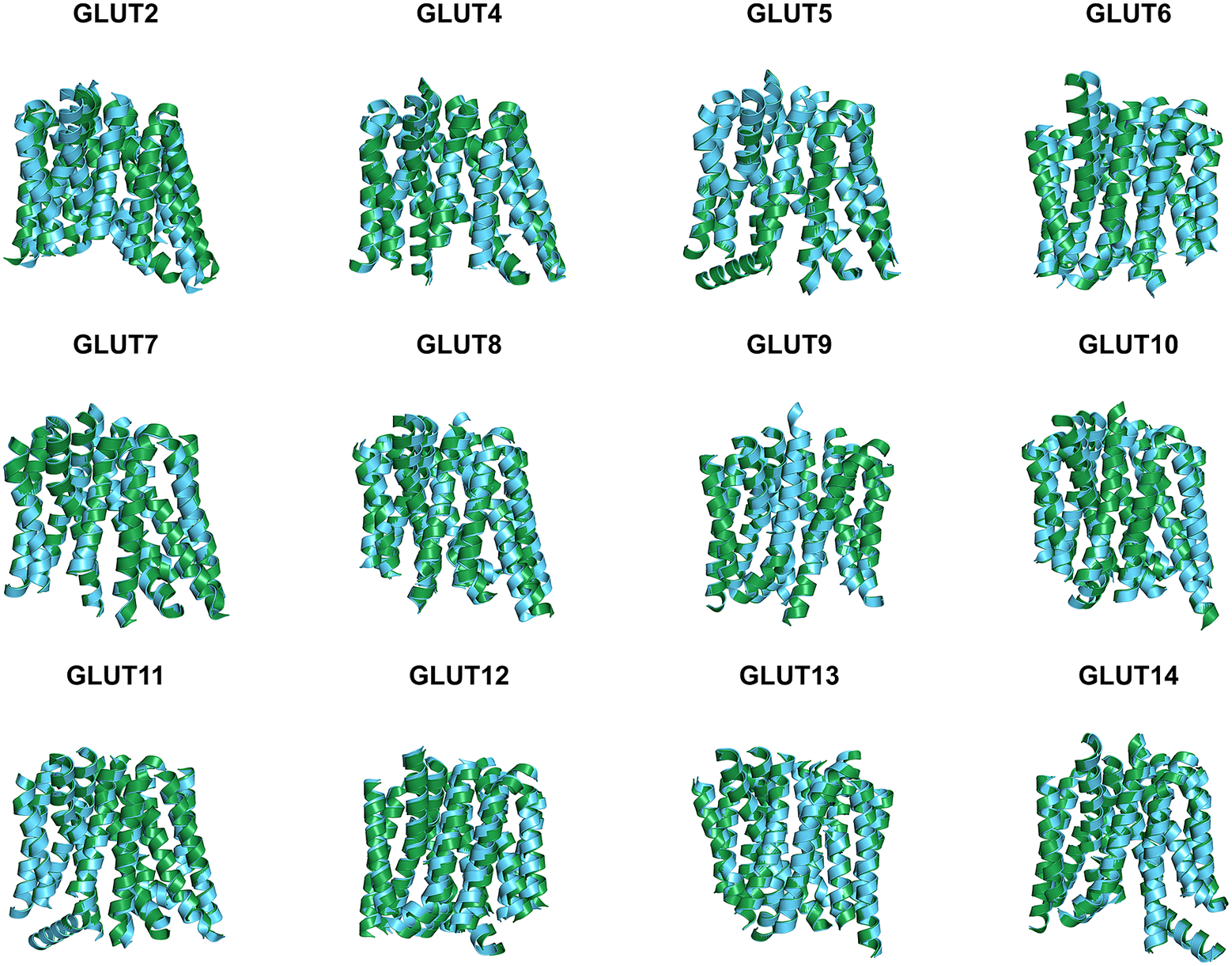
Fig. 4. Superposed AlphaFold2 predicted 12 native glucose transporters GLUT2, and from GLUT4 to GLUT14 and their QTY water-soluble variants. For clarity, large N- and C-termini are removed. The predicted native structures (green) and their water-soluble QTY variants (cyan). The RMSD in Å for the superposed structures are in (). (a) GLUT2 and GLUT2QTY (3.058 Å) (b) GLUT4 and GLUT4QTY (0.764 Å), (c) GLUT5 and GLUT5QTY (0.712 Å), (d) GLUT6 and GLUT6QTY (1.910 Å), (e) GLUT7 and GLUT7QTY (0.470 Å), (f) GLUT8 and GLUT8QTY (1.169 Å), (g) GLUT9 and GLUT9QTY (0.593 Å), (h) GLUT10 and GLUT10QTY (1.186 Å), (i) GLUT11 and GLUT11QTY (1.502 Å), (j) GLUT12 and GLUT12QTY (3.590 Å), (k) GLUT13 and GLUT13QTY (0.881 Å) and (l) GLUT14 and GLUT14QTY (2.720 Å). Please also see Table 3.
Analysis of the hydrophobic surface of native transporters and the water-soluble QTY variants
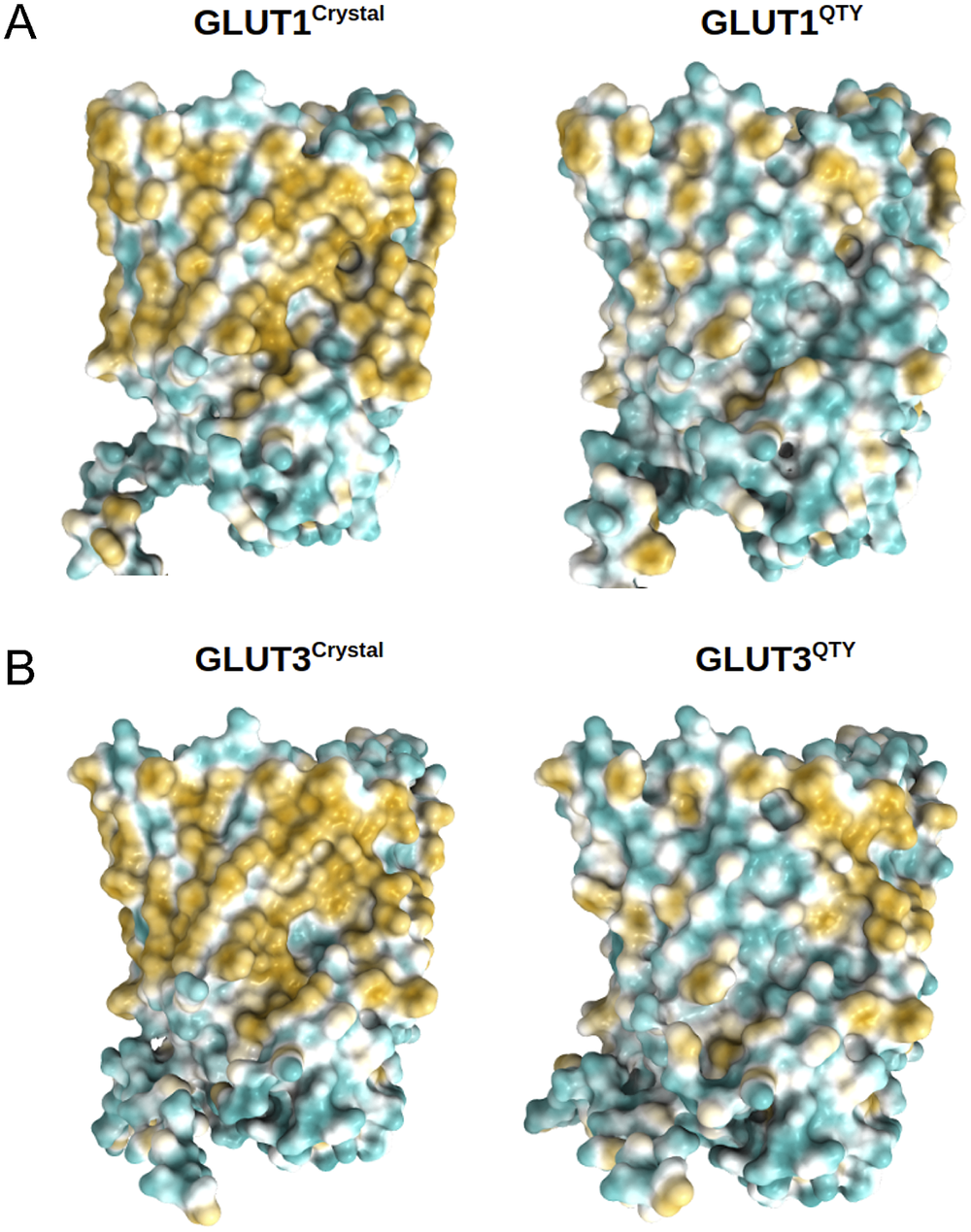
The native GLUTs are highly hydrophobic, especially in the 12TM helical domains, thus they are intrinsically water-insoluble and require detergents to solubilise them after removing them from the lipid bilayer membranes. Without the appropriate detergents, they immediately aggregate, precipitate and lose their biological functions.
The 12TM domains are directly embedded in the hydrophobic lipid bilayer so the hydrophobic side chains of amino acids leucine (L), isoleucine (I), valine (V) and phenylalanine (F) directly interact with the lipid molecules excluding water. Thus, they display highly hydrophobic patches on the 12TM domain (Figs 5 and 6 and Supplementary Fig. 1).
On the other hand, after systematic QTY replacement of hydrophobic amino acids L, I, V, F, with hydrophilic amino acids Q, T, Y, these hydrophobic patches become largely reduced (Figs 5 and 6). The QTY transformation from hydrophobic 12TM to hydrophilic 12TM did not significantly alter the alpha-helix structures. This would have been rather unexpected prior to obtaining the experimental results reported in recent publications (Zhang et al., Reference Zhang, Tao, Qing, Tang, Skuhersky, Corin, Tegler, Wassie, Wassie, Kwon, Suter, Entzian, Schubert, Yang, Labahn, Kubicek and Maertens2018; Qing et al., Reference Qing, Han, Skuhersky, Chung, Badr, Schubert and Zhang2019; Hao et al., Reference Hao, Jin, Zhang and Qing2020). However, that work demonstrated that QTY-designed chemokine receptors and cytokine receptors retained structure stability, integrity and ligand-binding function (Zhang et al., Reference Zhang, Tao, Qing, Tang, Skuhersky, Corin, Tegler, Wassie, Wassie, Kwon, Suter, Entzian, Schubert, Yang, Labahn, Kubicek and Maertens2018; Qing et al., Reference Qing, Han, Skuhersky, Chung, Badr, Schubert and Zhang2019, Reference Qing, Tao, Chatterjee, Yang, Han, Chung, Ni, Suter, Kubicek, Maertens, Schubert, Blackburn and Zhang2020; Hao et al., Reference Hao, Jin, Zhang and Qing2020; Tegler et al., Reference Tegler, Corin, Skuhersky, Pick, Vogel and Zhang2020).
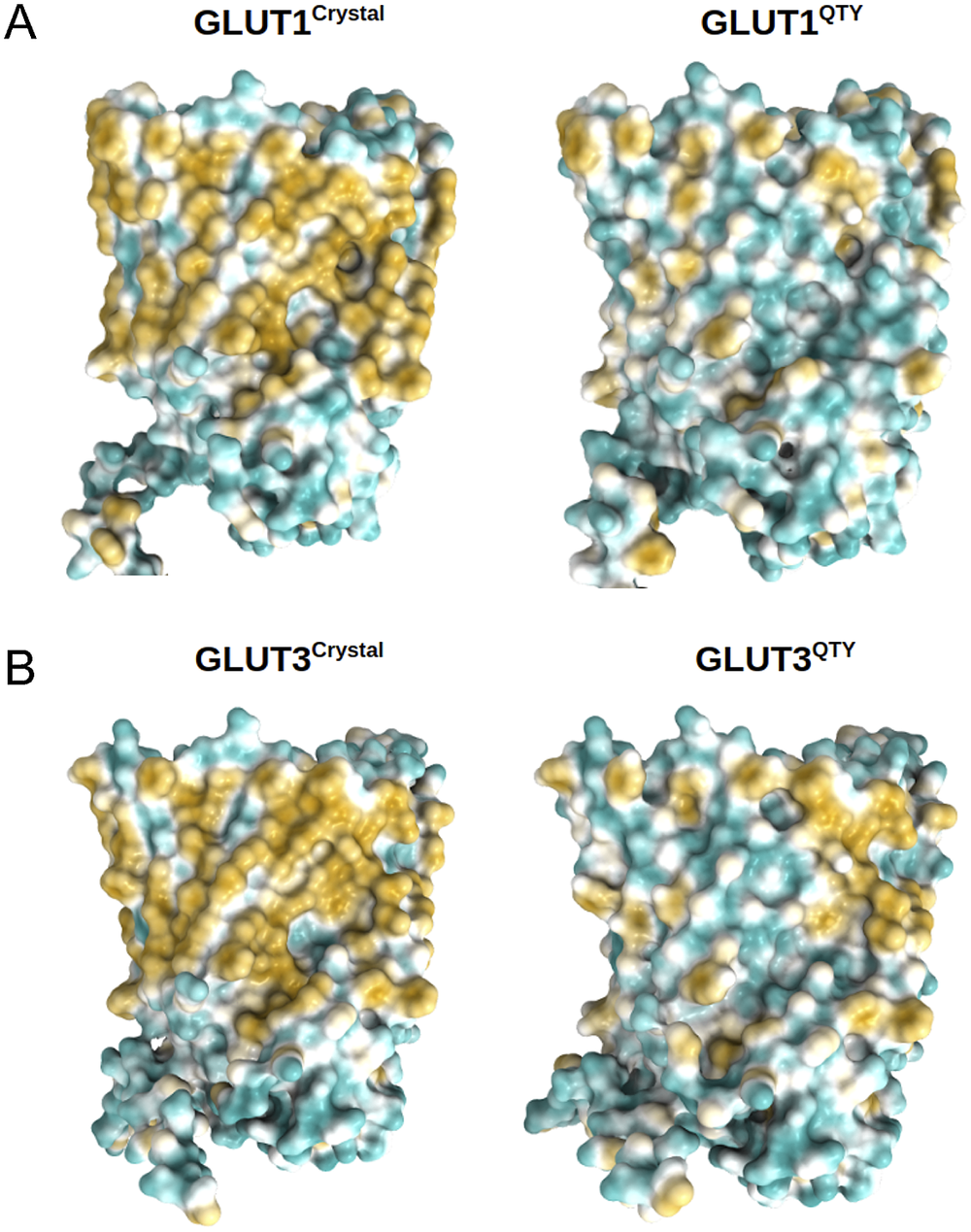
Fig. 5. Surface hydrophobic patch of crystal structures of native glucose transporters GLUT1 and GLUT3 and AlphaFold2 predicted water-soluble QTY variants. The native glucose transporters mostly expose hydrophobic residues leucine (L), isoleucine (I), valine (V) and phenylalanine (F) facing outside to the hydrophobic lipid bilayer in cell membrane. After replacing the L, I, V, F with polar amino acids, glutamine (Q), threonine (T) and tyrosine (Y), the surfaces are much less hydrophobic. The large surface hydrophobic patch (yellow colour) of the native receptors from X-ray crystal structures: (a) GLUT1Crystal and GLUT1QTY; (b) GLUT3Crystal and GLUT3QTY. The hydrophobic patch is significantly reduced on the transmembrane domains for the water-soluble QTY variants. These QTY variants become water-soluble without any detergent. The large N- and C-termini are removed for clarity of direct comparisons.
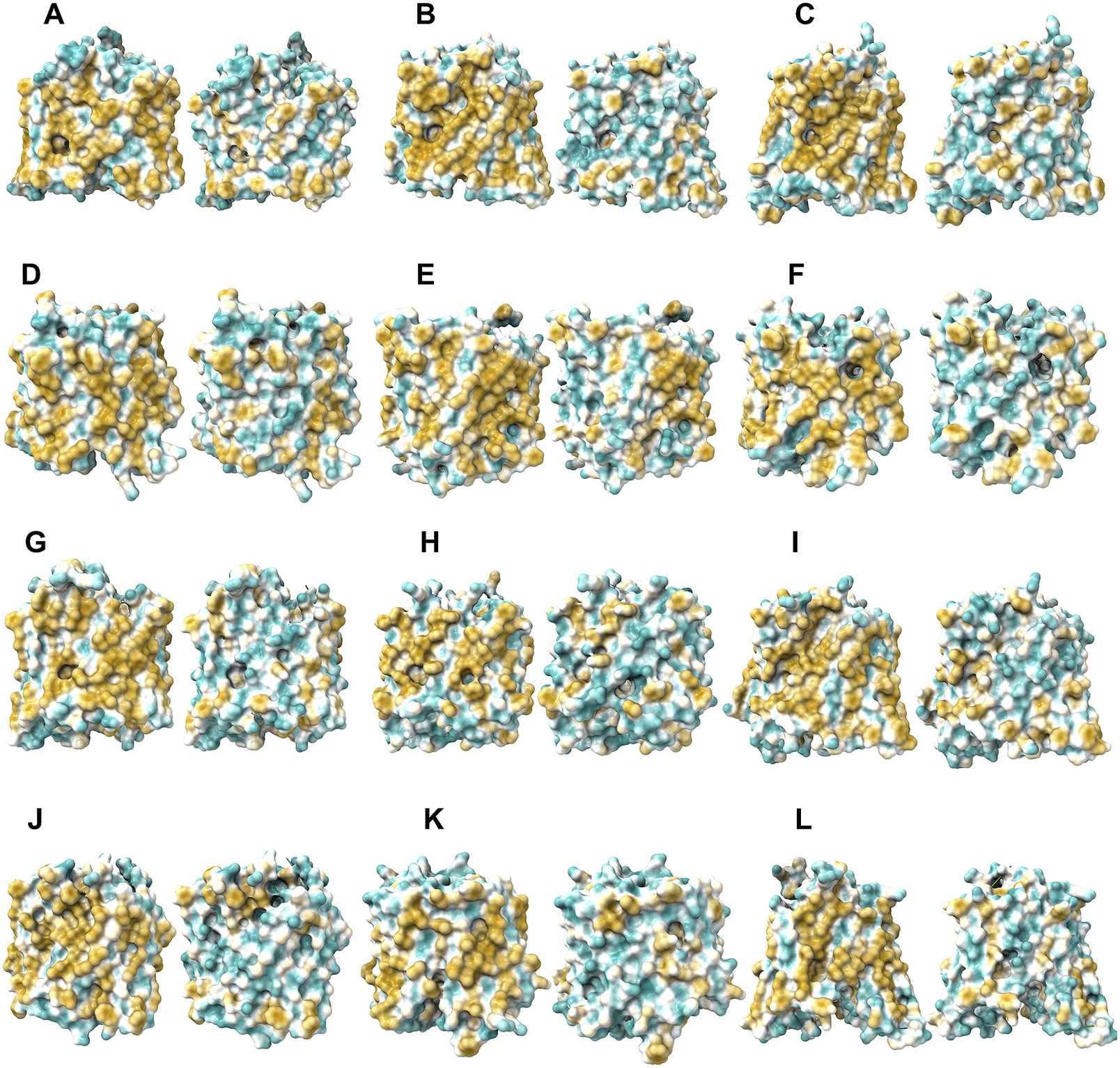
Fig. 6. Surface hydrophobic patch of AlphaFold2 predicted structures of native glucose transporters GLUT2, and from GLUT4 to GLUT14 and their water-soluble QTY variants. The pairwise of AlphaFold2 predicted native structures with large surface hydrophobic patch (yellowish colour), and the water-soluble QTY variant transporters (cyan colour): (a) GLUT2 and GLUT2QTY, (b) GLUT4 and GLUT4QTY, (c) GLUT5 and GLUT5QTY, (d) GLUT6 and GLUT6QTY, (e) GLUT7 and GLUT7QTY, (f) GLUT8 and GLUT8QTY, (g) GLUT9 and GLUT9QTY, (h) GLUT10 and GLUT10QTY, (i) GLUT11 and GLUT11QTY, (j) GLUT12 and GLUT12QTY, (k) GLUT13 and GLUT13QTY and (l) GLUT14 and GLUT14QTY. The N- and C-termini are removed for clarity of direct comparisons.
Nature has already evolved three distinct types of alpha-helices: (1) the hydrophilic alpha-helix such as found in haemoglobin and many other water-soluble enzymes and circulating proteins such as growth factors, cytokines and antibodies; (2) the hydrophobic alpha-helix such as in transporters and other integral transmembrane proteins found in G protein-coupled receptors, ion channels, photosynthesis systems and (3) amphiphilic alpha-helices with both hydrophilic and hydrophobic amino acid residues. These three types of alpha-helices have nearly identical molecular structures, irrespective of the hydrophobicity and hydrophilicity (Pauling and Corey, Reference Pauling and Corey1951; Branden and Tooze, Reference Branden and Tooze1999; Fersht, Reference Fersht2017). This insight is the structural basis of the QTY code.
AlphaFold2 predictions
For over six decades, structural biologists and protein scientists have sought to predict how proteins fold naturally and rapidly. We can now study protein structure in more detail in silico, and obtain previously unattainable protein structures, especially integral transmembrane proteins, through AlphaFold2 predictions, at least in the framework.
It is estimated that ~20–30% genes code for membrane proteins in most organisms (Krogh et al., Reference Krogh, Larsson, von Heijne and Sonnhammer2001) and ~25.86% of genes (5,139 among 21,416 annotated genes) in the human genome code for membrane proteins (Fagerberg et al., Reference Fagerberg, Jonasson, von Heijne, Uhlén and Berglund2010). However, determining the structure of even a single transmembrane protein is a daunting task, with many hurdles along the way, from gene expression, protein production, detergent selection, purification, to maintaining the stability, integrity and functionality to avoid aggregation. The determination of integral transmembrane protein structures lags far behind of those of water-soluble proteins.
We can now approach transmembrane structures computationally, using AlphaFold2 predictions, comparing the native structure with a proposed water-soluble QTY variant, and then express the water-soluble structure in vitro. We also modelled the glucose in its transporter channel (Fig. 7).
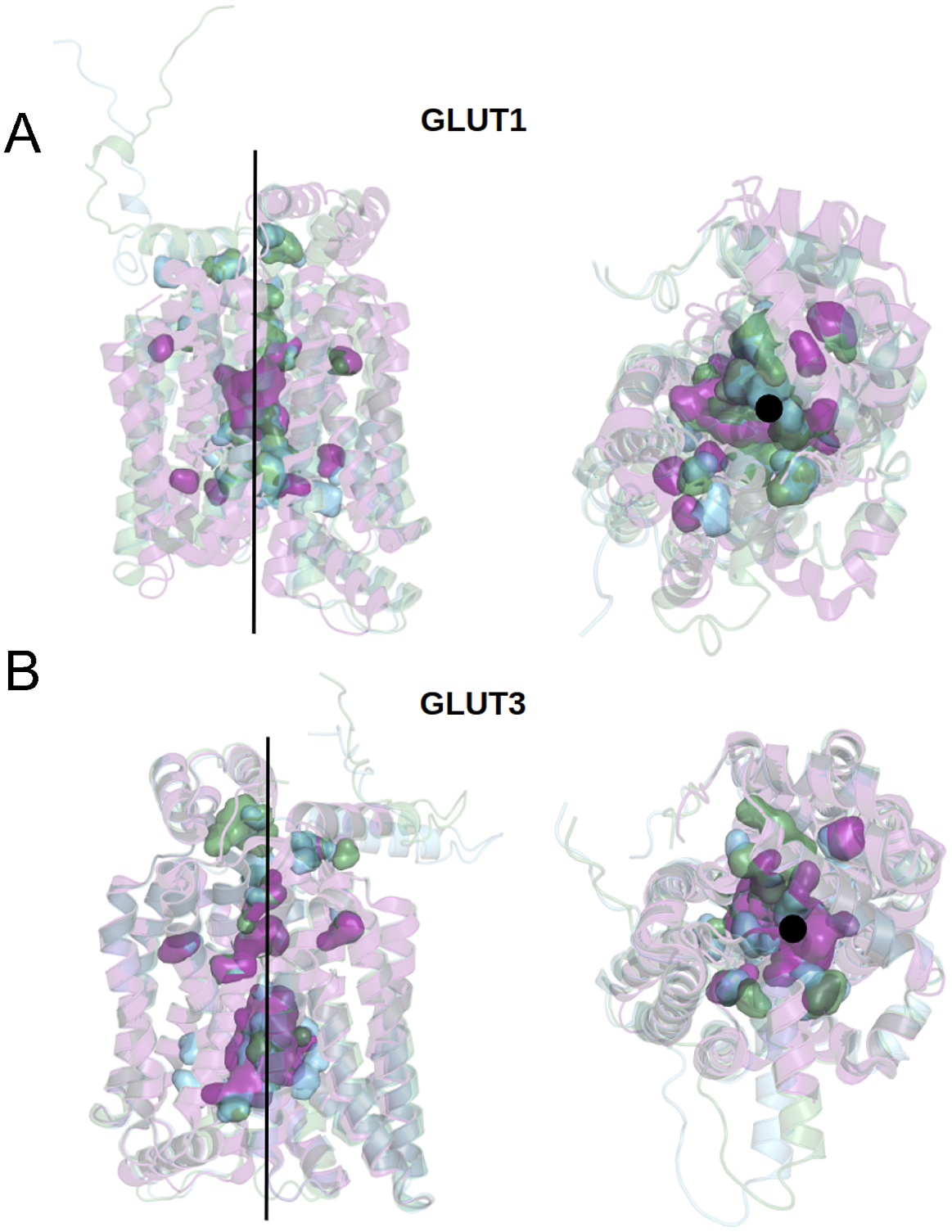
Fig. 7. Superposed structures of crystal, AlphaFold2 predicted native and QTY variants with the glucose channel. (a) GLUT1Crystal, GLUT1Native and GLUT1QTY, (b) GLUT3Crystal, GLUT3Native and GLUT3QTY. Black lines and dots and surface representation show the position of the glucose channel in three structures: crystal structures (magenta), AlphaFold2 predicted native (green) and AlphaFold2 predicted water-soluble QTY variants (cyan).
DeepMind AlphaFold’s team has already deposited ~ 1 million open protein structures in the EBI (https://alphafold.ebi.ac.uk). The number will continuously increase over time.
We previously used the AlphaFold2 accurate prediction tool to predict water-soluble chemokine receptors and compare them to the known experimentally determined structures (Skuhersky et al., Reference Skuhersky, Tao, Qing, Smorodina, Jin and Zhang2021). The speed and accuracy of AlphaFold2 predictions of our designed receptors is unprecedented. Instead of taking weeks or days to predict one structure, AlphaFold2 can compute a new structure in hours, or even minutes for smaller proteins. AlphaFold2 significantly accelerates studies of protein structures, stabilities, the design of new proteins, the discovery of new protein interactions, and perhaps new functions that were previously unknown through expensive and time-consuming experimental studies.
The location of glucose in the GLUT1native and GLUT1QTY variant
The crystal structure of native GLUT1 was determined with a glucose in the transporter channel (Deng et al., Reference Deng, Sun, Yan, Ke, Jiang, Xiong, Ren, Hirata, Yamamoto, Fan and Yan2015). It is interesting to know where the glucose is in the AlphaFold2 predicted water-soluble GLUT1QTY variant. As can be seen from superposed structures of native GLUT1 and GLUT1QTY variant (Fig. 7), the glucose still is in the same channel location as the native GLUT1native. Since both native and QTY variant superposed very well, the glucose in the channel is likely to be in the same location since glucose is highly hydrophilic and the QTY variant is unlikely to reduce its interaction with other hydrophilic amino acid residues.
Conclusion
Our study provides insight into the subtle differences between the hydrophobic and hydrophilic alpha-helices through systematically comparing experimentally determined structures GLUT1, GLUT3 and GLUT4 with AlphaFold2-predicted water-soluble QTY variant transporters, as well as AlphaFold2 predicted 11 native GLUT2 and from GLUT5 to GLUT14 and their QTY variants. Our study demonstrates the use of QTY-variant structures as a viable approach to modelling integral membrane proteins and other aggregated proteins. These GLUT water-soluble QTY variants may also be very useful for designing glucose sensing device and for drug discoveries.
Supplementary Materials
To view supplementary material for this article, please visit http://doi.org/10.1017/qrd.2022.6.
Acknowledgements
We thank Dr. Arthur Zalevsky for helpful discussions. We also thank Dorrie Langsley for English editing.
Author contributions
Conceptualization: S.Z.; Data curation: E.S.; Formal analysis: F.T.; Funding acquisition: D.J.; Investigation: F.T., R.Q., S.Y.; Methodology: E.S., F.T.; Project administration: S.Z.; Resources: D.J.; Software: AlphaFold2, E.S.; Supervision: S.Z.; Validation: E.S.; Visualisation: E.S.; Writing – original draft preparation: S.Z.; Writing – review and editing: E.S., F.T., R.Q., D.J., S.Y., S.Z. All authors have read and agreed to the published version of the manuscript.
Data availability statement
The AlphaFold2 predicted protein structures are at European Bioinformatics Institute (EBI, https://alphafold.ebi.ac.uk). The QTY code designed water-soluble variants are published in this article and at https://github.com/eva-smorodina/glucose-transporters.
Financial support
This work was primarily funded by Avalon GloboCare Corp.
Conflict of interest
Massachusetts Institute of Technology (MIT) filed several patent applications for the QTY code for GPCRs and OH2 Laboratories licenced the technology from MIT to work on water-soluble GPCR variants. However, this article does not study GPCRs. S.Z. is an inventor of the QTY code and has a minor equity of OH2 Laboratories shares. The funder Avalon GloboCare Corp had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.













 Open access
Open access
Comments
Comments to Author: This is an interesting paper reporting analysis of mutated in silico hydrophobic sequences into hydrophilic ones in transmembrane helical segments using the QTY tool in analysis of glucose transporters that are implicated in various cancers. The authors have used the AlphaFold2 predictions for potential water-soluble QTY variants of these membrane transporters by mutating the transmembrane helices making them hydrophilic. The predicted structures of mutated glucose transporters GLUT2, and GLUT4 to GLUT14 were compared with known structures of GLUT1 (PDB: 6THA) and GLUT3 (PDB: 4ZW9) revealing remarkable similarity.
This is a quite important approach that has potential vital impact on further studies glucose transporters saving significant amount of time and finances in analysis of function of this group of proteins.
It would be important ifthe authors will address some questions:
It was not clear how the solubility of the potential hydrophilic glucose transporters will change their functions. It would be good to model their interactions with glucose. It was not clear if the cancer related overexpression/upregulation glucose transporters were related to their incorporation into membranes or changes in their transport of glucose. Highly possible that this info has been published in some papers and that must be verified and analysed in silico.Plausibly that these channels by which glucose is transported though the membrane were changed, but this issue was not analysed.
What is importance of the isoelectric focusing point of glucose transporters?Is it different in the cancer cells? Hope this is known from previous publications.
Additional comments
It would be much easier for a reader to have a table where GLUT 1-14 would be listed and in this table related cancers, availability of a structure, and references would be indicated.This part of introduction is rather difficult to read and recapitulate the most important facts.It was not clear if these glucose transporters were mutated in cancerous cell and were not able to be incorporated into membranes or just did not function. That would be good to know.
It would be good to see in the figure where is the glucose channel located within the proteins obtained by X-ray.It would be important to see the same areas in silico mutants.
It would be recommended to replace throughout the entire MS the word "superimpose" to "superpose"
Superposed - Place (something) on or above something else, esp. so that they coincide. Used mostly in scientific or mathematical contexts. To place one geometric figure on top of another in such a way that all common parts coincide.
Superimpose-To place an object over another object, usually in such a way that both will be visible. Typically related to 2D images, one does not worry if they are not coinciding.
Figure 3. It would be good if the authors will check the orientations GLUT1 and GLUT3 in this figure. It seems that views (front and side) are swapped for GLUT3 compared to the view of GLU1.The last panel of GLUT3 (Fig3b) seems to be rotated in plane and in a different orientation with respect to GLUT1 as well. It is strongly recommended to indicate where are the N - and C-termini and the angles between different views. Nothing is shown in green. Please check the legend.