INTRODUCTION
Avian influenza is a disease of major importance for poultry. It is caused by type A influenza viruses which can infect a wide variety of animal species including many wild bird and poultry species, swine and humans [Reference Alexander1]. It is a very diverse virus and currently 16 haemagglutinin (HA) and nine neuraminidase (NA) subtypes have been discovered in birds [Reference Fouchier2]. Most HA and NA subtypes can be found in many possible combinations in wild water fowl, which are the virus's natural reservoir [Reference Olsen3].
Avian influenza viruses (AIV) are typically classified in two pathotypes; highly pathogenic avian influenza (HPAI) and low pathogenic avian influenza (LPAI), based on the symptoms developed in chickens. Highly pathogenic avian influenza viruses (HPAIVs) (formerly known as bird flu or fowl plague) cause severe illness and high mortality in chickens. These virulent phenotypes have only been observed in H5 and H7 serotypes. Infection of chickens with low pathogenic avian influenza viruses (LPAIVs) can be asymptomatic or can cause mild to severe respiratory sickness and/or general illness which may or may not be associated with secondary infections. LPAI is found in HA serotypes 1–16 (including H5 and H7) [Reference Swayne and Pantin-Jackwood4]. The circulation of H5 and H7 LPAIVs in poultry populations can lead to mutations resulting in the insertion of basic amino acids at the HA0 cleavage site. Since this causes the virus to become highly pathogenic, these two subtypes are classified as notifiable viruses [5].
Direct or indirect contact between poultry and wild birds can lead to the introduction of AIV in the poultry sector. Therefore, location in the vicinity of wild-bird breeding grounds or major flyways, the possibility of close direct contact between wild birds and domestic birds, or breaches in biosecurity measures may increase the risk of introducing H5/H7 AIV into poultry holdings [Reference Koch and Elbers6, Reference Welby7]. Such events in which an H5/H7 LPAIV was introduced into the poultry sector and subsequently transformed into a highly pathogenic form of the virus have been observed during the 1983–1984 outbreak in Pennsylvania, the 1993–1994 outbreak in Mexico and the 1999 outbreak in Italy [Reference Capua8–Reference Horimoto10].
Precise estimation of whether an H5/H7 LPAI outbreak poses a risk of giving rise to an outbreak of HPAI is impossible. However, it is generally assumed that the wider the circulation of H5/H7 LPAIV in a population, the higher the probability of an HPAIV emerging [Reference Alexander11]. Therefore, studying the transmissibility of LPAIV isolates that have been isolated in poultry and wild birds can provide more insight in this process. A better understanding of the transmission of H5 and H7 LPAIVs is needed for controlling the circulation of these viruses and thereby reducing the risk of them becoming highly pathogenic.
Quantification of disease transmission can be done by using the basic reproduction ratio (R 0), which is essentially the average number of susceptible individuals that are infected by one typical infectious individual during its entire infectious period in a fully susceptible population [Reference Diekmann, Heesterbeek and Metz12]. This definition implies that an infection may spread throughout a susceptible population if R 0>1, and otherwise may die out if R 0<1 [Reference Velthuis13].
The survival of AIVs in water, poultry litter and on surfaces has been demonstrated, indicating that fomites may contribute in AIV transmission [Reference Reis14–Reference Tiwari16]. Moreover, poultry housing systems have been suggested to affect transmission of AIV in the initial stage of an outbreak [Reference Tsukamoto17] and Chen et al. [Reference Chen18] have suggested the spread of an H5N1 HPAIV between ducks is compromised by housing the animals on a grid. Considering the role of these factors in the circulation of AIV in poultry can greatly increase our understanding of the transmission of the disease.
In this study, the transmissibility of two poultry originated LPAIVs and one duck originated LPAIV was assessed in a series of transmission experiments involving direct contact between chickens. To provide more insight into the process of LPAIV transmission, the viruses that were selected for the present study have different infective properties for poultry, more specifically regarding their tissue tropism (oropharyngeal vs. cloacal replication). In addition, experiments were conducted pairwise on different floor systems to assess the impact of virus properties and floor system on virus transmission.
METHODS
Viruses
Three LPAIVs were used in the present study. LPAIV H5N2 A/Ch/Belgium/150VB/99 was isolated by the Veterinary and Agrochemical Research Institute (VAR) in 1999. This isolate was obtained from chickens in a smallholder flock where ∼100 chickens and 20 ducks were held. The chickens experienced 10% mortality associated with clinical signs such as depression, diarrhoea and respiratory distress. This virus was probably introduced by the purchase of 10 chickens from a local market 10 days earlier [Reference Alexander19]. A second egg passage of this virus was used for inoculation of the animals.
A second LPAIV, H7N1 A/Ch/Italy/1067/v99, was isolated from chickens by the Istituto Zooprofilattico Sperimentale (IZS) during the 1999 LPAI epidemic in northeastern Italy [Reference Capua8]. A fourth egg passage of this virus was used for inoculation of the animals.
The third LPAIV used in this study, H5N3 A/Anas platyrhynchos/Belgium/09–884/2008, was isolated in Belgium from the cloacal swab of a wild mallard duck that was sampled as part of a long-term wild-bird monitoring programme that ran from August 2008 to April 2009 [Reference Van20]. A second egg passage of this virus was used for inoculation of the animals.
Animals
All animal experiments were conducted on specific pathogen free (SPF) chickens, delivered by Lohmann-Valo (Germany). The animals were housed in biosafety level-3 isolators (type: HM1500, Montair Process Technology B.V., The Netherlands) from the day of hatching until the end of the experiment. The isolators have a floor surface of 1·2 m2 and the internal volume measures 0·9 m3. A negative air pressure of 45 ± 5 m3/h was maintained during the entire course of the experiment. Each animal experiment was conducted under the authorization and supervision of the Biosafety and Bioethics Committee at VAR, following national and European regulations.
Experimental design
Three transmission experiments were conducted, each one using one of the three viruses described above. Each transmission experiment comprised two (for the H5N2 and H7N1 viruses) or one (for the H5N3 virus) trial(s) in which virus transmission was studied in two separate groups of SPF chickens, hereafter referred to as subtrials or housing groups (Fig. 1). Animals from these two subtrials were housed in different isolators, with a different floor system. In one subtrial, animals were housed on a grid flooring, which allowed droppings to pass through. In the other subtrial, the floor of the isolator was covered with plastic on which ∼1·5 kg of litter (wood shavings, Agrospan Houtkrullen, Vividerm, Belgium) was spread. To reduce other variations between the two floor systems as much as possible, both subtrials were conducted at the same time, with the same lot of SPF chickens and the same inoculum. The second trial of a transmission experiment was a repetition of the first trial.

Fig. 1 [colour online]. Schematic illustration of the design of transmission experiments conducted in this study. Experiments with H5N2 A/Ch/Belgium/150VB/99 and H7N1 A/Ch/Italy/1067/v99 comprised two separately conducted trials. The experiment with H5N3 A/Anas platyrhynchos/Belgium/09-884/2008 comprised only one trial. Each trial included two subtrials which were simultaneously conducted in different isolators; in one isolator, animals were housed on a grid floor; in the other isolator the floor was covered with litter. In each isolator, six seeders (red diamonds) and six contacts (green ovals) were housed.
In each subtrial, six SPF chickens were oculo-nasally inoculated at age 4–6 weeks with a 106 EID50/dose virus solution. These animals are hereafter referred to as seeders. One day after inoculation, six naive SPF chickens, hereafter referred to as contacts, were introduced into the isolator. This is the reference time point used in this study and will be referred to as 0 day post-exposure (dpe). Oropharyngeal and cloacal swabs were taken from the seeders from 0 dpe until 9 dpe. For the contacts, oropharyngeal and cloacal swabs were taken from 1 dpe until 10 dpe. At 14 and 21 dpe, blood samples were taken from all animals.
Sample handling
After sampling, the oropharyngeal and cloacal swabs were immediately submerged in a falcon tube filled with 1·5 ml storage medium containing brain-heart infusion broth enriched with antibiotics (BHI+AB) (106 U/l penicillin G, 2 g/l streptomycin, 1 g/l gentamycin sulfate, 66 ml/l kanamycine sulfate 100×). Drinking water was sampled by collecting 1·5 ml drinking water and pouring it into 1·5 ml doubly concentrated BHI+AB medium, to yield the same concentration of medium and antibiotics in drinking-water samples as that present in swab samples. Swabs and water samples were subsequently stored at −80°C, for further analysis. Sera from blood samples were stored at −20°C.
Detection and quantification of viral RNA in samples using one-step real-time reverse transcription–polymerase chain reaction (rRT-PCR)
Viral RNA (vRNA) was semi-automatically extracted from 50 μl thawed sample material using a KingFisher magnetic particle processor and the MagMax™ AI/ND-96 Viral RNA kit (Ambion Inc., USA), according to the manufacturer's protocol. The Quantitect Probe RT–PCR kit (Qiagen GmBH, Germany) was used to prepare a total of 25 μl reaction volume (containing 2 μl purified RNA) for amplification of matrix gene in a Biosystems 7500 real-time PCR cycler (Applied Biosystems, Belgium) [Reference Van21]. A series of 1:10 dilutions of synthetic matrix RNA was run simultaneously in each rRT–PCR run to calculate the number of vRNA copies in each sample. Then, a series of 1:10 dilutions of the stock solution of each virus was analysed to create a standard curve from which EID50 equivalents per ml (EID50 eq/ml) sample medium of each sample could be calculated. Results were finally expressed as EID50 eq/ml storage medium or drinking water. Samples with an RNA concentration <100·0 EID50 eq/ml were considered negative. The selection of samples to be analysed was based on data needed for assessment of transmission parameters.
Serology
Serum samples were tested for antibodies directed towards the viral nucleoprotein with IDScreen influenza A antibody competition ELISA kit (IDvet, France). All tests were conducted according to the manufacturer's instructions. In the data analysis, serum samples with a doubtful result were considered positive.
Statistical analysis
Reproduction ratios were estimated using the susceptible–infectious–recovered (SIR) model, as described in Velthuis et al. [Reference Velthuis13]. In a SIR model, fully susceptible individuals that are in contact with infectious individuals can either stay susceptible or become infectious and finally recover from infection. The number of susceptible, infectious and recovered animals for each time period as well as the number of new cases (animal passing from exposed to infectious state) observed in the same time period were recorded. Animals were considered infected when anti-AIV antibodies were present at either 14 dpe, 21 dpe or both.
Basic reproduction ratios were estimated using two different methods. In the first method, R 0 was estimated using following formula:
 $$R_0 = \beta /\alpha. $$
$$R_0 = \beta /\alpha. $$In this formula, transmission rate (β) was obtained following a Generalized Linear Model (GLM) developed in SAS v. 9.2 and described in Velthuis et al. [Reference Velthuis13, Reference Gonzales22, Reference Gonzales23]. The parameter log β was estimated by modelling the number of new infections upon contact, using the offset function ln(IΔt/N), and the complementary log-log function. A back-transformation of log β was required to obtain β. The recovery rate (α) was calculated as the arithmetic mean of the individual infectious periods of all infected contact animals. The 95% confidence intervals (CI) and P values used to determine whether the results were significant or not were those obtained for the parameter log β. The goodness of fit of the GLM was assessed using Akaike's Information Criterion. For the calculation of transmission parameters with this method, contact animals were considered infected when seroconversion was demonstrated at 14 dpe and/or 21 dpe and when virus shedding >101·3 EID50 eq/ml was observed at least once. The same cut-off value was maintained for determining the infectious periods.
In the second method, values for R 0 were estimated according to the Final Size (FS) model, using the maximum-likelihood estimator [Reference Gonzales22, Reference Dewulf24–Reference van der Goot27]:
 $$R_0 = \max \prod\nolimits_{i = 1}^n {{\rm Prob}(x_i{\cdot}R_0 |N{\cdot}S_0{\cdot}I_0 ).} $$
$$R_0 = \max \prod\nolimits_{i = 1}^n {{\rm Prob}(x_i{\cdot}R_0 |N{\cdot}S_0{\cdot}I_0 ).} $$Confidence intervals were constructed symmetrically around the estimate of R 0, in accordance with the method described by Bouma et al. [Reference Bouma28]. For this method, animals were considered infected if seroconversion was demonstrated at either 14 dpe or 21 dpe.
RESULTS
Transmission of H5N2 A/Ch/Belgium/150VB/99
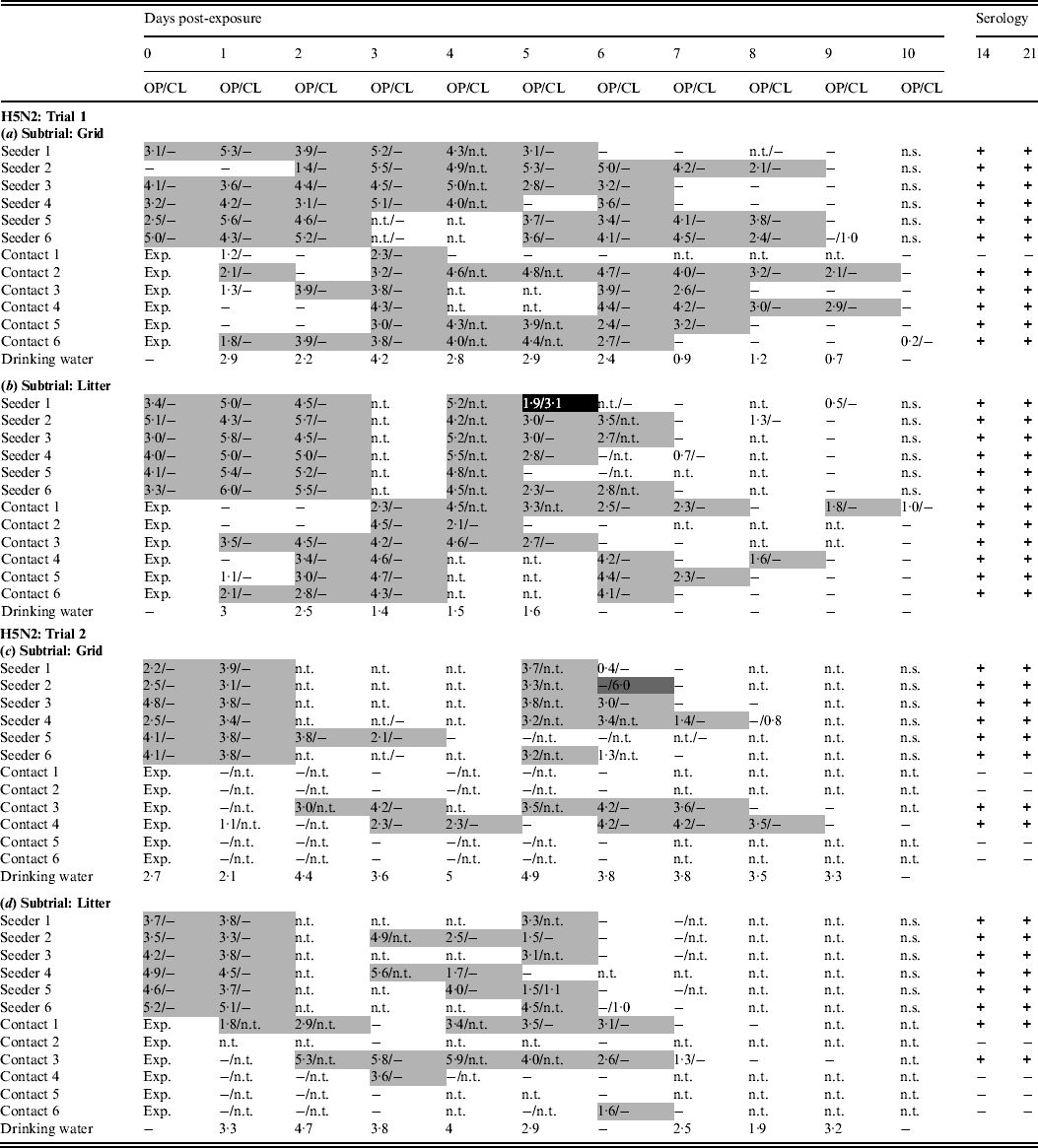
In trial 1, the seeders shed virus from the day after inoculation (0 dpe) until at least 4 dpe, except for one animal in the grid subtrial, which commenced virus shedding at 2 dpe (Table 1a). Virus shedding was nearly always via the oropharynx. Cloacal virus shedding was demonstrated in only one inoculated animal, in the litter subtrial, at 5 dpe (Table 1b). In both subtrials, virus shedding was observed in all contact animals. In the grid subtrial, one contact animal was found positive by rRT–PCR on two separate days and did not seroconvert. All other contact animals in this subtrial shed virus for several consecutive days and seroconverted (Table 1a). In the litter subtrial, one contact animal was also found positive by rRT–PCR for only 2 days; however, in this case these were consecutive days, larger EID50 eq/ml were demonstrated in these swabs and this animal seroconverted. All other contact animals in the litter subtrial demonstrated virus shedding for several consecutive days and seroconverted (Table 1b). Viral RNA was found in drinking water until 8 dpe for the grid subtrial and 5 dpe for the litter subtrial.
Table 1. Results for transmission experiments with H5N2 A/Ch/Belgium/150VB/99 LPAIV
OP, Oropharyngeal; CL, cloacal.
Virus shedding is expressed as log10 EID50 eq/ml storage medium for OP and CL swabs. Light grey squares = OP swab with virus concentration >101·3 EID50 eq/ml and CL swab ⩽101·3 EID50 eq/ml or not tested; dark grey squares = CL swab with virus concentration >101·3 EID50 eq/ml and OP swab ⩽101·3 EID 50 eq/ml or not tested; black squares = OP and CL swabs both containing a virus concentration >101·3 EID50 eq/ml. Virus in drinking water is expressed as log10 EID50 eq/ml. Immune response at 14 and 21 days post-exposure as determined by ELISA test. +, Positive; ±, doubtful; −, negative; n.t., not tested; n.s., no sample; exp., exposure. (a) Trial 1: subtrial 1; (b) trial 1: subtrial 2; (c) trial 2: subtrial 1; (d) trial 2: subtrial 2.
In trial 2, all seeders from both subtrials shed virus from 0 dpe until at least 3 dpe. Virus shedding was practically always via the oropharynx and cloacal virus shedding was only rarely observed. In the grid subtrial, virus shedding was observed in two contact animals, both of which were positive from 1 or 2 dpe until 7 or 8 dpe. Both these animals were the only ones from this group to seroconvert (Table 1c). In the litter subtrial, two contact animals showed a similar pattern of virus shedding and were also the only ones to seroconvert. In contrary to the group housed on a grid, two more contact animals from the litter housing group were found positive by rRT–PCR for 1 day, although without seroconverting (Table 1d). Drinking water contained vRNA from 0 or 1 dpe until 9 dpe.
In general, transmission of H5N2 A/Ch/Belgium/150VB/99 was different in both trials. Despite virus shedding in seeders being similar in all groups for both trials, fewer contact animals became infected in trial 2 than in trial 1. Virus transmission leading to seroconversion always occurred during the 3 days following introduction of the contact animals and no cloacal virus shedding was observed during this period.
Transmission of H7N1 A/Ch/Italy/1067/v99
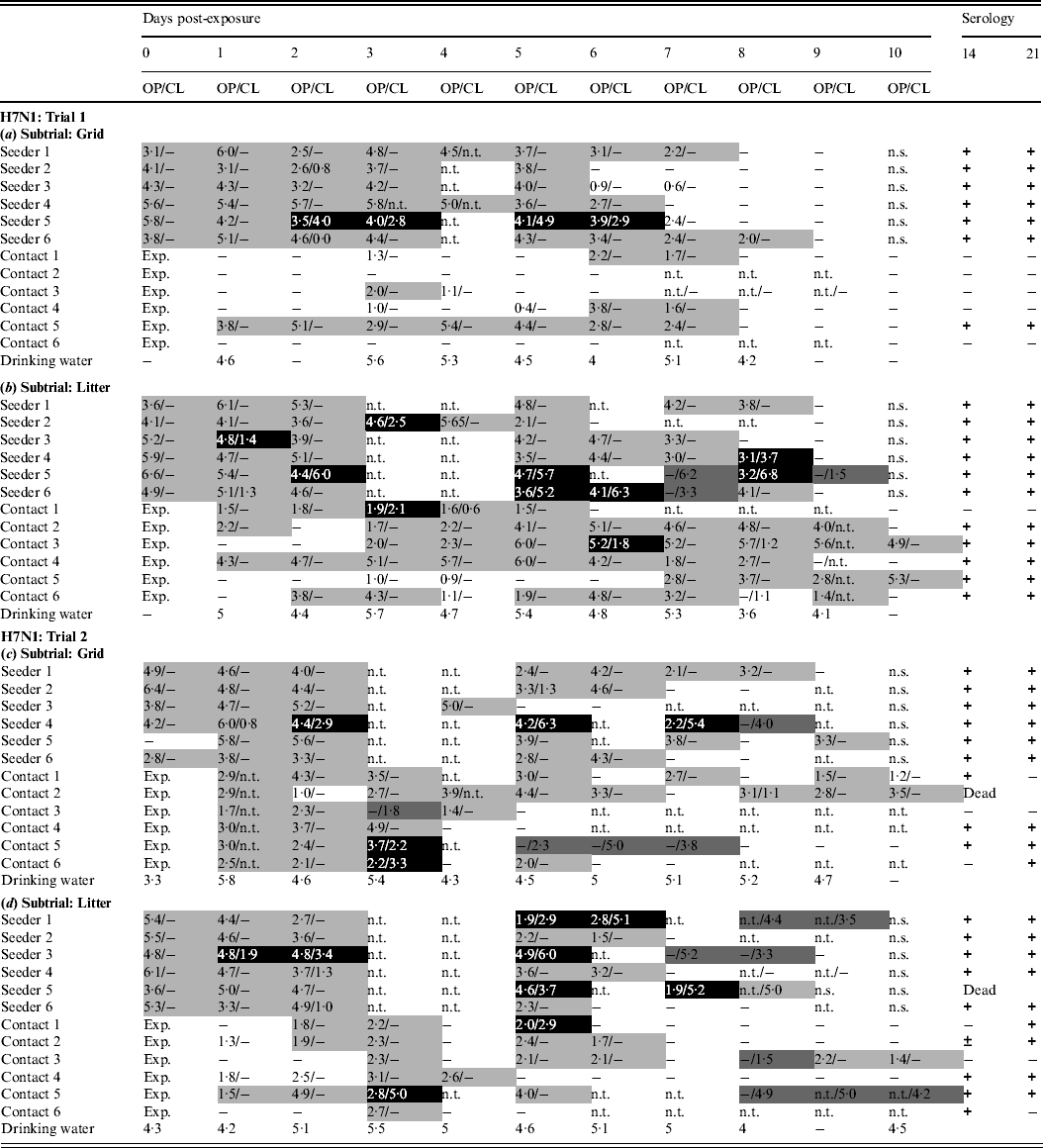
In trial 1, the seeders shed virus from the day after inoculation until at least 5 dpe. Oropharyngeal virus shedding preceded combined virus shedding via the oropharynx and cloaca in three animals of the grid subtrial (Table 2a) and five animals of the litter subtrial (Table 2b). In the grid subtrial, only one contact animal seroconverted. This animal shed virus for 7 consecutive days. Three other contact animals were found positive by rRT–PCR on several occasions but did not seroconvert (Table 2a). In the group housed on litter, all animals were found positive by rRT–PCR throughout most of the experiment. Cloacal virus shedding was observed in two contact animals and five contact animals seroconverted (Table 2b). Drinking water contained vRNA throughout most of the experiment.
Table 2. Results for transmission experiments with H7N1 A/Ch/Italy/1067/v99 LPAIV
OP, Oropharyngeal; CL, cloacal.
Virus shedding is expressed as log10 EID50 eq/ml storage medium for OP and CL swabs. Light grey squares = OP swab with virus concentration >101·3 EID50 eq/ml and CL swab ⩽101·3 EID50 eq/ml or not tested; dark grey squares = CL swab with virus concentration >101·3 EID50 eq/ml and OP swab ⩽101·3 EID50 eq/ml or not tested; black squares = OP and CL swabs both containing a virus concentration >101·3 EID50 eq/ml. Virus in drinking water is expressed as log10 EID50 eq/ml. Immune response at 14 and 21 days post-exposure as determined by ELISA test. +, Positive; ±, doubtful; −, negative; n.t., not tested; n.s., no sample; exp., exposure. (a) Trial 1: subtrial 1; (b) trial 1: subtrial 2; (c) trial 2: subtrial 1; (d) trial 2: subtrial 2.
In trial 2, all seeders shed virus until at least 4 dpe. One inoculated animal from the grid subtrial was negative by rRT–PCR at 0 dpe, but was found positive 1 day later. Cloacal virus shedding was observed in two seeders from the grid subtrial (Table 2c) and five seeders from the litter subtrial (Table 2d). In the grid subtrial, all contact animals were rRT–PCR positive from 1 dpe until at least 3 dpe. Then, one animal ceased virus shedding, two animals were found positive on two more days while the other three animals were found positive by rRT–PCR until at least 7 dpe. No blood samples were available from one contact animal from the grid subtrial that died at 10 dpe from a non-AIV related cause. For the remaining five contact animals from this group, four animals seroconverted at 14 dpe and/or 21 dpe. The one contact animal that did not seroconvert was found positive by rRT–PCR for four consecutive days but did not shed large amounts of virus. For the litter subtrial, one contact animal was found shedding large doses of virus via the oropharynx and cloaca from 1 dpe until the end of the trial and seroconverted. The other contact animals, however, were positive at several times, but the amounts of virus shed were clearly smaller. Nevertheless, four of these animals were found positive on serology (Table 2d). Viral RNA was demonstrated in drinking water from 0 dpe until 9 or 10 dpe.
In both trials, the number of seeders that shed viruses via the cloaca was higher in the groups housed on litter compared to the groups housed on a grid, despite virus shedding at 0 dpe being highly comparable for all subtrials. In trial 1, the virus was transmitted more successfully in the group housed on litter. However, no difference regarding the number of contacts becoming infected was observed between the two housing groups in trial 2.
Transmission of H5N3 A/Anas platyrhynchos/Belgium/09-884/2008
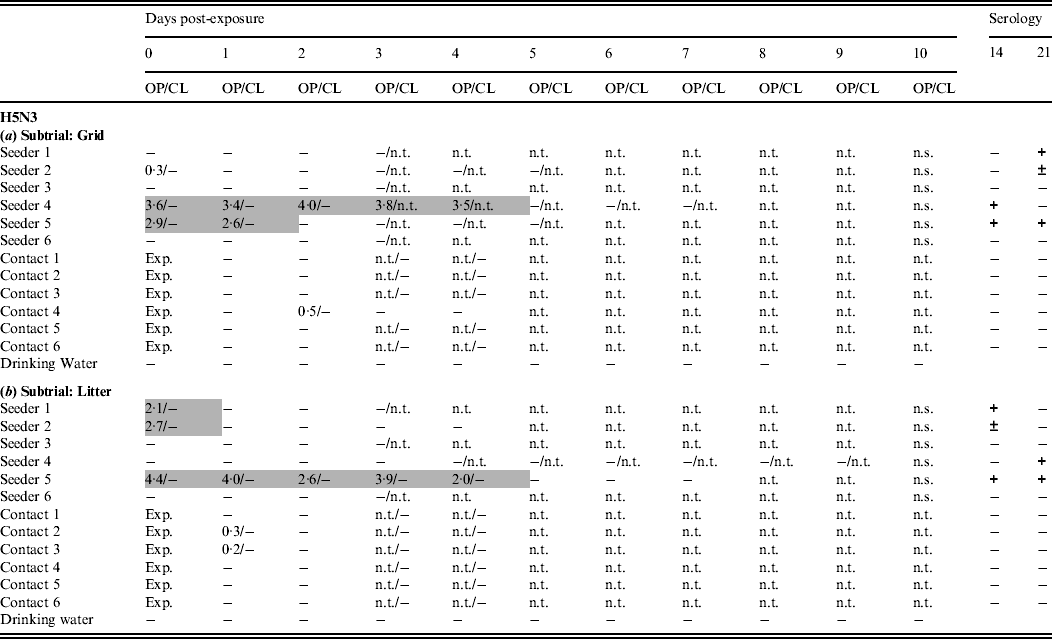
Only one trial was conducted with this virus. In the grid subtrial, only two seeders shed virus through the oropharynx for ⩾2 days. A third animal was found slightly positive by rRT–PCR at 0 dpe. Seroconversion was demonstrated in four seeders (Table 3a). In the litter subtrial, virus shedding by seeders was comparable; one animal shed virus for 5 days while two other animals were found positive at 0 dpe only. Seroconversion was also seen in four seeders (Table 3b). In the grid subtrial, one contact animal was found slightly positive by rRT–PCR at 2 dpe and none seroconverted (Table 3a). In the litter subtrial, two contact animals were found slightly positive by rRT–PCR. Again, none of the contact animals seroconverted (Table 3b). In both subtrials, no virus was detected by rRT–PCR in any of the drinking-water samples.
Table 3. Results for transmission experiments with H5N3 A/Anas platyrhynchos/Belgium/09-884/2008 LPAIV
OP, Oropharyngeal; CL, cloacal.
Virus shedding is expressed as log10 EID50 eq/ml storage medium for OP and CL swabs. Light grey squares = OP swab with virus concentration >101·3 EID50 eq/ml and CL swab ⩽101·3 EID50 eq/ml or not tested; dark grey squares = CL swab with virus concentration >101·3 EID50 eq/ml and OP swab ⩽101·3 EID50 eq/ml or not tested; black squares = OP and CL swabs both containing a virus concentration >101·3 EID50 eq/ml. Virus in drinking water is expressed as log10 EID50 eq/ml. Immune response at 14 and 21 days post-exposure as determined by ELISA test. +, Positive; ±, doubtful; −, negative; n.t., not tested; n.s., no sample; exp., exposure. (a) Subtrial 1; (b) subtrial 2.
Quantification of virus transmission
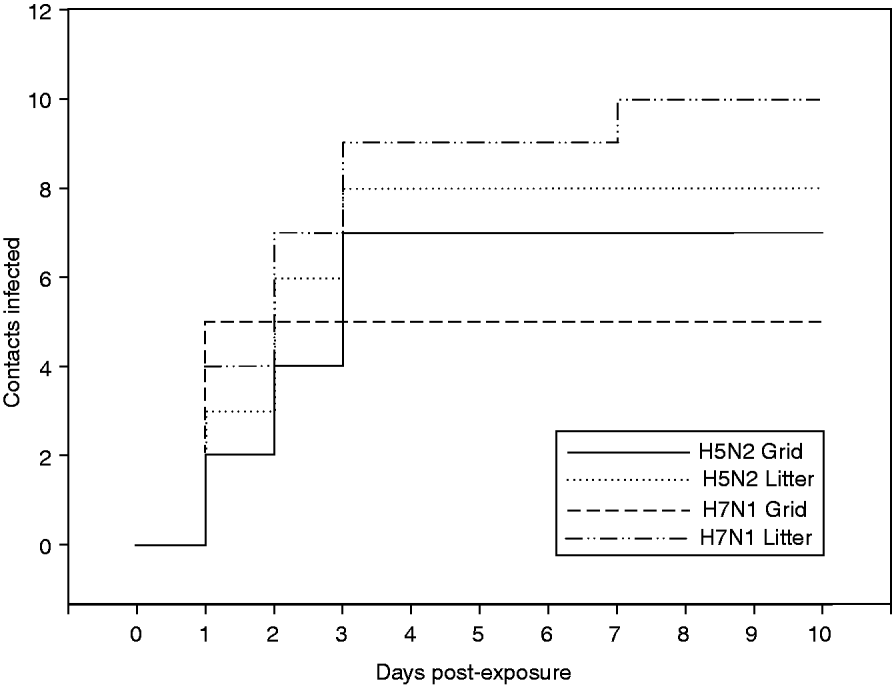
Figure 2 shows the evolution of the number of contact-infected animals per time unit. For the H5N2 experiment, all infections on both floor systems occurred within 3 dpe. For the H7N1 experiment, the graphs for the two floor systems have different courses (Fig. 2).
Fig. 2. Evolution of the number of contact-infected animals for experiments with H5N2 A/Ch/Belgium/150VB/99 and H7N1 A/Ch/Italy/1067/v99 conducted on two different floor systems.
Quantification of virus transmission was initially conducted for each subtrial separately (results not shown). Since results of subtrials with the same flooring and virus were not significantly different, these subtrials were combined in order to increase the precision of the study.
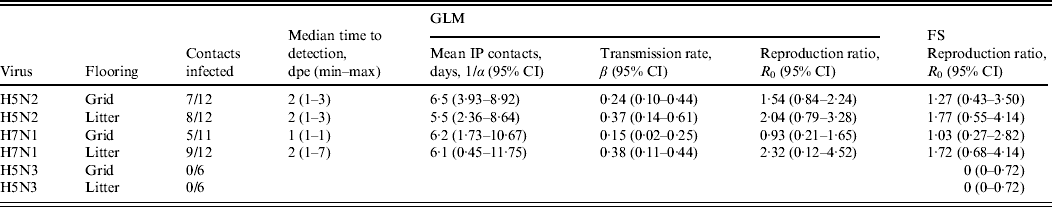
Table 4 summarizes the results for combined transmission experiments and the reproduction ratios estimated according to GLM and the FS method. Transmission of the H5N2 and H7N1 viruses was successful on all floor types. Table 4 shows that the point estimates of the basic reproduction ratios for the H5N2 experiment are >1 for both floor systems, suggesting this virus is capable of spreading through a susceptible population. However, the 95% CI calculated for both floor systems was large and not significantly greater than 1. Both point estimates for the two floor systems evaluated in this study do not differ much and both CIs largely overlap, suggesting that there is no important difference in virus transmission between the two floor systems. For the H7N1 experiment, values for R 0 are larger for the subtrials conducted on litter than on grid. However, in these trials the 95% CIs also largely overlap. Since none of the contact animals in the experiments with H5N3 A/Anas platyrhynchos/Belgium/09-884/2008 seroconverted, the R 0 for this virus could not be obtained with the GLM.
Table 4. Quantification of transmission for combined experiments with A/Ch/Belgium/150VB/99 (H5N2), A/Ch/Italy/1067/v99 (H7N1) and A/Anas platyrhynchos/Belgium/09-884/2008 (H5N3) on each floor system, according to the Generalized Linear Model (GLM) and the Final Size (FS) model
CI, Confidence interval; IP, infectious period; dpe, days post-exposure.
Basic reproduction ratios estimated using the FS model are comparable to values for R 0 estimated by the GLM and mostly have larger 95% CIs.
DISCUSSION
In the present study, the impact of two parameters, virus strain and flooring, on virus transmission was assessed in a series of transmission experiments involving direct contact between inoculated and susceptible SPF chickens. The three selected LPAIVs had different infective characteristics for the host species. The impact of flooring on virus transmission was assessed by dividing each trial in two subtrials that were conducted simultaneously and differed only in the floor system used. Subtrials where animals were housed on a grid were chosen to mimic cage housing or the housing of animals on raised floors on which droppings do not accumulate, while subtrials in which the floor of the isolator was covered with litter were used to mimic conditions in an all litter housing system. Several parameters such as infectious period, time of infection and basic R 0 were determined to quantify the impact of these parameters on virus transmission.
To the best of our knowledge, only a few studies have quantified transmission of H5 and H7 LPAIVs between chickens in laboratory experiments. Van der Goot et al. [Reference van der Goot27] estimated R 0 for A/Chicken/Pennsylvania/21525/83 H5N2 LPAIV and Gonzales et al. [Reference Gonzales22, Reference Gonzales23] estimated R 0 for A/Chicken/Netherlands/2006 H7N7 LPAIV, A/Turkey/Italy/1067/99 H7N1 LPAIV and observed no transmission of A/Turkey/Italy/2369/2009 H5N7 LPAIV. It is generally assumed that viruses that are well adapted to a certain bird species replicate easily in this species and are shed in large amounts [Reference Swayne and Slemons29, Reference Spekreijse30], giving rise to a large infection pressure in the environment, which has been suggested to determine the incidence and the course of infection [Reference Bouma31].
The chicken originated H5N2 virus selected in this study proved to be successfully transmitted between SPF chickens. Preliminary studies have shown that H5N2 A/Ch/Belgium/150VB/99 has a strong infectious potential for SPF chickens and is shed almost exclusively via the oropharynx (G. Claes, unpublished data). The present study demonstrates that cloacal virus shedding among seeders was only seen in a minority of animals and only occurred near the end of the infectious period. Since most contact-infected birds were already shedding virus before that time, it can be said that transmission of this virus occurred through oropharyngeal virus shedding by infectious animals and that aerosols, which are proven to be important in LPAIV transmission [Reference Tsukamoto17, Reference Okamatsu32, Reference Yee33], play an important role in the transmission of this virus. The differences in transmission parameters between subtrials with this virus conducted on grid or litter flooring were insignificant and might rather be attributed to biological variation than to an impact of the floor system.
Transmission of H7N1 A/Ch/Italy/1067/v99 from inoculated SPF chickens to susceptible SPF chickens occurred in all trials. Regarding the impact of floor system on transmission of this virus, we observed: (i) more contact animals seroconverting, (ii) more seeders presenting cloacal virus shedding, and (iii) infection of contact animals throughout the entire course of the experiment for the litter housing groups. These observations suggest an impact of the floor system on the transmission of this virus. Moreover, the joint R 0 estimated for the subtrials conducted on grid flooring showed that less transmission occurred in this housing condition, compared to the subtrials conducted on litter. However, since the CIs of the reproduction ratios were strongly overlapping, it is impossible to conclude, based on the current results, whether this difference is a true biological difference or rather due to coincidence.
This result might be linked to the substantial affinity of H7N1 A/Ch/Italy/1067/v99 for intestinal epithelia of infected animals. Our results agree with Marché et al. [Reference Marché, Lambrecht and van den Berg34], where cloacal shedding of H7N1 A/Ch/Italy/1067/v99 occurred after a short period (2 or 3 days) of virus replication in the oropharynx or trachea. Therefore, after an initial transmission of virus via aerosols, transmission between animals housed on litter may have been aided by the accumulation of infectious faeces. By contrast, housing of animals on a grid provided a constant discharge of faeces and thereby impeded a further increase of infection pressure.
Regarding the assessment of reproduction ratios for this virus, the 10-day time-frame used to monitor virus shedding in the animals appears to be too short. Virus shedding >101·3 EID50 eq/ml was still observed in some animals at 9 or 10 dpe, suggesting that virus shedding still continued at 11 dpe and that the mean infectious period is underestimated. Basic reproduction ratios for H7N1 A/Ch/Italy/1067/v99 were smaller than basic reproduction ratios that have previously been estimated by Gonzales et al. [Reference Gonzales22] for a similar virus from the same LPAIV outbreak. However, in both studies the criteria that were used to define the status of individuals within the SIR model were different.
Next to the results presented in this study, both H7N1 and H5N2 strains used have genetic evidence of adaptation to poultry as the length of the stalk of their NA is reduced. Details can be found in the EpiFlu databank (isolate ID: Epi_isl_74837 [35]). Interestingly, both these poultry-adapted LPAIVs have different virus shedding patterns. Based on the results from our study, the intestinal tropism of LPAIVs should not be considered as the only mechanism for poultry adaptation. Whether this statement counts for only certain AIV subtypes or whether it represents an intrinsic characteristic for each LPAIV is still to be explained. Other studies might suggest that this replication tropism might be subtype specific as the characteristics of the H5N2 A/Ch/Bel/150VB/99 were highly comparable to the characteristics observed by van der Goot et al. for H5N2 A/Chicken/Pennsylvania/21525/83 LPAIV [Reference van der Goot27]. Both strains have the same distinct affinity for the upper respiratory tract, are hardly shed via the cloaca and have very similar estimates for R 0.
There is no evidence that H5N3 A/Anas platyrhynchos/Belgium/09-884/2008 was transmitted to the contact animals. A preliminary study has proven this virus to be of low infectivity for poultry since <50% of inoculated SPF chickens established virus shedding after oculo-nasal inoculation with a 106 EID50 viral dose. Moreover, virus shedding has proved to be very short, mainly oropharyngeal and generally weak (G. Claes, unpublished data). A study by Van Borm et al. [Reference Van20], which focused on the genetic properties of this and related viruses, indicated no evidence of adaptation to poultry, such as deletions in the NA stalk region. Therefore, the virus was incapable of successfully infecting enough inoculated animals to build up the infective pressure required for transmission and/or was too quickly eliminated by the host's immune response to allow virus shedding. The latter probably explains why seroconversion was observed for two seeders in which virus shedding was not detected. In analogous experiments, Gonzales et al. [Reference Gonzales23] observed no transmission of H5N7 A/Turkey/Italy/2369/2009 LPAIV between layer chickens. These findings corroborate the hypothesis that an adaptation step would be needed for the spread of similar viruses in chicken flocks.
Several fomites have been suggested to contribute to the spread of AIV. For example, it is well known that AIVs can remain infectious in water reservoirs for a considerable amount of time [Reference Rohani15, Reference Stallknecht36–Reference Nazir40]. Some recent animal experiments on AIV transmission have found virus titres in drinking water high enough to cause virus transmission [Reference Achenbach and Bowen41]. Therefore, daily water samples from experiments performed in this study were analysed for the presence of vRNA. Our results show drinking-water samples from experiments with H5N2 and H7N1 contained large amounts of vRNA, sometimes ranging as high as 105 EID50 eq/ml. Additional tests have demonstrated that viruses from these samples were viable (data not shown). Since the drinking water was refreshed each time after sampling, it can be assumed that the quantities of vRNA found in the drinking water was a good indication of the virus shedding or virus charge in the isolator at each time interval. Indeed, for seven out of eight experiments where vRNA was demonstrated in the drinking water, the first day on which virus shedding was demonstrated in most of the contact animals coincides with the day on which peak concentrations of vRNA were found in the drinking water. However, whether the increase of vRNA in the drinking water is either the cause or consequence of infection of contact animals is difficult to assess as a possible contamination of oropharyngeal swabs (animal drinking just before sampling) cannot be excluded.
Conducting experiments under laboratory conditions inevitably leads to an artificial environment. Fewer variables are thus involved, which enhances the reproducibility of experiments. On the other hand, the atmosphere created by controlled temperature, air flow and relative humidity may differ from what is appropriate in the field and the sustainability of virus in faeces or on surfaces such as the grid floor or wood shavings may be altered [Reference Lowen42, Reference Guan43]. Similarly, the susceptibility of SPF chickens to LPAIVs may be different from that of conventional chicken breeds.
Virus transmission was evaluated according to the SIR model [Reference de Jong and Kimman44]. Basic reproduction ratios of this model were estimated using the FS method and GLM. Estimation of reproduction ratios using the FS method is independent of a latency period. By contrast, the GLM method does take the latency period into account since each time unit of the experiment is considered instead of just the final state [Reference Velthuis26]. Because the GLM makes use of more input data, the 95% CIs of R 0 estimated by this method are generally smaller than when the FS method is used.
In transmission experiments that use the SIR model, the outcome is largely dependent on the criterion that is used to define the status of an animal and on the diagnostic assay that is used to detect infection [Reference Dewulf25, Reference Comin45]. Indeed, for many pathogens that cause no or only minor symptoms in diseased animals, it is sometimes difficult to categorically demonstrate freedom from disease in the flock. Since this is certainly the case for LPAIV, a wide variety of criteria defining an infected contact animal in LPAIV transmission studies are used in the literature. Isolation of virus from oropharyngeal or cloacal swabs from animals is traditionally regarded as a sign of infection [Reference Li46–Reference Westbury, Turner and Amon48] while some studies have included serology in their data analysis [Reference Gonzales22, Reference Gonzales23, Reference van der Goot27]. In recently conducted LPAIV transmission experiments, rRT–PCR has frequently been used to determine infection in contact animals [Reference Gonzales23, Reference Bertran49–Reference Yee, Cardona and Carpenter51]. In our study, we assumed that oropharyngeal swabs can be positive by rRT–PCR because the animal picked up viruses from the environment just before sampling, which can lead to a positive rRT–PCR result without the animal actually being infected. Therefore, we used the more conservative approach that assumes that a true virus replication leads to seroconversion and shedding of fairly large doses of virus. For this reason, only animals that seroconverted were considered infectious and a stricter rRT–PCR cut-off value than simply the limit of detection of this method was used to assess the moment of infection, the infectious period and the moment of recovery. Our choice of a 101·3 EID50 eq/ml cut-off value was based on a small analysis of the results that had shown that, when using this cut-off value, most animals that are considered infected are indeed positive by both rRT–PCR and the ELISA test, thereby reducing the number of animals that are positive by rRT–PCR but negative by ELISA or vice versa as much as possible (data not shown).
In this study, we have demonstrated that the floor system in poultry barns might have an impact on transmission of LPAIVs that are replicated in the intestinal tract of infected animals. However, since the observed effect was rather small, this environmental factor should not be considered as a critical factor for LPAIV transmission and other factors should be considered influential as well. Indeed, transmission of LPAIVs must be considered as a highly complex event and since all these environmental factors act upon the infection pressure, their impact on transmission, and thus the risk of the virus becoming highly pathogenic, should not be underestimated.
ACKNOWLEDGEMENTS
The authors thank Anouar Nadi, Marc Boschmans, Christophe Delgrange and Marc van den Broeck for their technical and laboratory assistance. We also thank Dr Ilaria Capua (Istituto Zooprofilattico Sperimentale delle Venezie, Legnaro, Padova, Italy) for providing the H7N1 virus used in this study. This work was supported by Belgian Contractual Research (Flutree, grant RF6217).
DECLARATION OF INTEREST
None.