



There is an increasing disparity in the prevalence of cigarette smoking between the general population and people with severe mental illness such as bipolar disorder.Reference Dickerson, Schroeder, Katsafanas, Khushalani, Origoni and Savage1,Reference Jackson, Diaz, Lopez and Leon2 A meta-analysis including worldwide studies showed that smoking rates are much higher in people with bipolar disorder than in the general population (OR = 3.5, 95% CI 3.39–3.54).Reference Jackson, Diaz, Lopez and Leon2 This is important since in people with bipolar disorder smoking is associated with negative physical and mental health outcomes compared with non-smoking.Reference Icick, Gard, Barde, Carminati, Desage and Guillaume3,Reference Depp, Bowie, Mausbach, Wolyniec, Thornquist and Luke4 Smoking has been suggested as a risk factor for developing severe mental illness, including psychosisReference MacCabe5 and – to a lesser extent – bipolar disorder.Reference Talati, Bao, Kaufman, Shen, Schaefer and Brown6–Reference Slyepchenko, Brunoni, McIntyre, Quevedo and Carvalho9 There is also an alternative explanation, namely that people with bipolar disorder initiate smoking or increase existing smoking. In the latest genome-wide association study (GWAS) of smoking, smoking initiation and heaviness and bipolar disorder were not found to be genetically correlated.Reference Liu, Jiang, Wedow, Li, Brazel and Chen10 Smoking and schizophrenia are genetically correlated.Reference Liu, Jiang, Wedow, Li, Brazel and Chen10 Nevertheless, smoking rates in bipolar disorder are also very high. A recent study showed, furthermore, that bipolar I disorder and schizophrenia and bipolar II disorder and major depressive disorder are highly correlated.Reference Ruderfer, Ripke, McQuillin, Boocock, Stahl and Pavlides11 Moreover, previous findings support a causal relationship between smoking and other severe mental illness (e.g. schizophrenia and major depressive disorder).Reference Wootton, Richmond, Stuijfzand, Lawn, Sallis and Taylor12 It is therefore of interest to investigate the causal relationship between smoking and bipolar disorder. So far, very few observational studies have been used to investigate this relationship.Reference Talati, Bao, Kaufman, Shen, Schaefer and Brown6 Observational results can be limited in their ability to determine causality, particularly because of residual confounding and reverse causation. Mendelian randomisation (MR) is a method that aims to overcome these limitations. Therefore, our aim was to investigate the direction and causal nature of the relationship between smoking and bipolar disorder using a bidirectional MR approach. On the basis of the current evidenceReference Talati, Bao, Kaufman, Shen, Schaefer and Brown6,Reference Thomson, Berk, Dodd, Rapado-Castro, Quirk and Ellegaard7,Reference Slyepchenko, Brunoni, McIntyre, Quevedo and Carvalho9 and a strong genetic correlation between bipolar disorder and schizophrenia,Reference Ruderfer, Ripke, McQuillin, Boocock, Stahl and Pavlides11 we hypothesised that smoking is a causal risk factor for bipolar disorder. On the basis of conflicting clinical findingsReference Heffner, Strawn, DelBello, Strakowski and Anthenelli8 and the fact that smoking initiation often precedes the onset of bipolar disorder symptoms, we hypothesised that the liability to bipolar disorder is not a causal risk factor for smoking.
Method
Mendelian randomisation
In this MR study, genetic variants are used as instrumental variables and the plausibility of the findings depends on assumptions. The three key assumptions of MR are: (a) the multiple genetic variants associate with the risk factor of interest; (b) there are no unmeasured confounders of the associations between the genetic variants and outcome; and (c) the genetic variants affect the outcome only through their effect on the risk factor.Reference Aad, Abbott, Abdallah, Abdinov, Aben and Abolins13 Pleiotropic effects refer to the effects of a genetic variant on multiple biological pathways, affecting the outcome either via the pathway under investigation, known as horizontal pleiotropy, or affecting other traits through the risk factor, known as vertical pleiotropy.Reference Davey Smith and Hemani14 Horizontal pleiotropy is problematic as it violates the third key assumption.
Data sources and genetic instruments
In this study, multiple genetic variants that collectively explain some of the variance in the risk factors are used to investigate support for causal inferences with clinical outcomes. For smoking and bipolar disorder, genome-wide significant single nucleotide polymorphisms (SNPs) (P <5 × 10−8) from the most recently available GWASs were used as genetic instruments. No overlap existed in GWAS samples used for the exposure and outcome instruments.
Data availability
For smoking initiation, we used summary data from the GWAS and Sequencing Consortium of Alcohol and Nicotine use (GSCAN) GWAS as an exposure and outcome variable.Reference Liu, Jiang, Wedow, Li, Brazel and Chen10 GSCAN summary data about smoking heaviness and cessation from the GSCAN GWAS were used as an outcome variable only.Reference Liu, Jiang, Wedow, Li, Brazel and Chen10 For lifetime smoking, we used summary data from the GWAS in the UK Biobank for the exposure and outcome variables.Reference Wootton, Richmond, Stuijfzand, Lawn, Sallis and Taylor12
For bipolar disorder, genome-wide significant SNPs (P < 5 × 10−8) from the most recently available GWAS were used as genetic instrument for the exposure.Reference Stahl, Breen, Forstner, McQuillin, Ripke and Trubetskoy15 When bipolar disorder was the outcome, we used the publicly available GWAS summary data made available by Psychiatric Genomics Consortium (https://www.med.unc.edu/pgc/results-and-downloads/).
Smoking behaviour
Smoking initiation
For smoking initiation, we used summary data from a GWAS conducted by GWAS and Sequencing Consortium of Alcohol and Nicotine use (GSCAN) as an exposure and outcome variable. The GSCAN GWAS identified 378 independent significant loci from a sample of 1 232 091 individuals of European ancestry: these loci explained 2.3% of the variance in smoking initiation.Reference Liu, Jiang, Wedow, Li, Brazel and Chen10 Data from the 23andMe subsample (n = 599 289) were excluded when smoking initiation was the outcome because genome-wide summary statistics were not publicly available for the 23andMe subsample. Smoking initiation (yes/no) was defined as having smoked >100 cigarettes over the course of your life, smoked every day for at least a month or ever smoked regularly. The GSCAN GWAS provided effect sizes calculated from z-values weighted by the average prevalence across all studies. The effect sizes for smoking initiation as the outcome are therefore presented as betas and correspond to the increase of one standard deviation in prevalence of smoking initiation.Reference Liu, Jiang, Wedow, Li, Brazel and Chen10
Smoking heaviness and cessation
To assess causal effects of smoking heaviness and smoking cessation on the risk for bipolar disorder, the analyses would have to be stratified into groups of smokers and non-smokers. However, in the current two-sample MR study this was not possible owing to a lack of data on smoking in the bipolar disorder GWAS. Therefore, summary data about smoking heaviness and cessation from the GSCAN GWAS were used as an outcome variable only (supplementary material, available at https://doi.org/10.1192/bjp.2019.202).Reference Liu, Jiang, Wedow, Li, Brazel and Chen10
Lifetime smoking
The recently developed lifetime smoking index is a measure that applies to all individuals (i.e. ever and never smokers), and among smokers it captures smoking severity, length of exposure and cessation. This instrument does not require stratification and is therefore the best proxy to investigate a ‘dose–response’ effect on the risk for bipolar disorder. Therefore, we used lifetime smoking summary data from the GWAS in the UK Biobank.Reference Wootton, Richmond, Stuijfzand, Lawn, Sallis and Taylor12 This GWAS of lifetime smoking identified 126 independent genome-wide significant SNPs in a sample of 462 690 individuals of European ancestry.Reference Wootton, Richmond, Stuijfzand, Lawn, Sallis and Taylor12 It has been validated in an independent sample, where the 126 SNPs explained 0.36% of the variance in lifetime smoking, and against positive control disease outcomes (e.g. lung cancer).Reference Wootton, Richmond, Stuijfzand, Lawn, Sallis and Taylor12
Bipolar disorder
For bipolar disorder as an exposure, we used summary statistics on genome-wide significant SNPs (P <5 × 10−8) from the most recent GWAS, comprising 20 352 cases from Europe, North America and Australia and 31 358 controls of European descent.Reference Stahl, Breen, Forstner, McQuillin, Ripke and Trubetskoy15 This GWAS found 30 independent, genome-wide significant loci in the combined meta-analysis of the discovery GWAS and follow-up samples. The combined analyses also used 9412 cases and 137 760 controls from follow-up samples. The polygenic risk score, including all measured genetic variants, accounted for about 8% of the variance in diagnosis. In this data-set, three clinically recognised subtypes of bipolar disorder were present: bipolar I disorder (n = 14 879), bipolar II disorder (n = 3421) and schizoaffective disorder bipolar type (n = 977). When bipolar disorder was the outcome, we used the GWAS summary data made available by the Psychiatric Genomics Consortium with a similar number of bipolar disorder cases and controls (https://www.med.unc.edu/pgc/results-and-downloads/).Reference Stahl, Breen, Forstner, McQuillin, Ripke and Trubetskoy15
Statistical analyses
We used the MR-Base PackageReference Hemani, Zheng, Elsworth, Wade, Haberland and Baird16 in R version 3.5.117 for Windows to harmonise SNP information and conduct bidirectional summary-level MR analyses between smoking and bipolar disorder. The magnitude of the causal effect can be estimated by dividing the ‘genetic variant-outcome’ association by the ‘genetic variant-risk factor’ association.Reference Davies, Holmes and Davey Smith18 Results were compared across the following methods: inverse-variance weighted (IVW), MR-Egger,Reference Bowden, Smith and Burgess19 MR-Egger simulation extrapolation method (SIMEX),Reference Bowden, Del Greco, Minelli, Smith, Sheehan and Thompson20 weighted median,Reference Bowden, Smith, Haycock and Burgess21 weighted modeReference Hartwig, Smith and Bowden22 and MR robust adjusted profile score (MR-RAPS)Reference Zhao, Jingshu, Gibran, Jack and Small23 (see supplementary material for a comparison of each method and their assumptions). A consistent direction of effect across all of these methods strengthens causal evidence, as each method makes different assumptions about pleiotropy.Reference Lawlor, Tilling and Davey Smith24 Cochran's Q (for IVW) and Rucker's Q (for MR-Egger) statistics were therefore calculated to investigate heterogeneity between SNP causal effects and thus potential evidence of horizontal pleiotropy. Evidence of directional pleiotropy (whereby pleiotropic effects are biasing the estimates in the same direction) was also assessed using the MR-Egger intercept. The difference between Cochran's Q and Rucker's Q statistics (Q′) was used to assess the extent to which MR-Egger was a better fit to the data than the IVW method. The intercept from MR-Egger provides a formal test for the presence of directional pleiotropy (whereby pleiotropic effects are biasing the estimates in the same direction). The validity of the MR-Egger method was also evaluated using the weighted and unweighted regression dilution I 2GX statistic.Reference Bowden, Del Greco, Minelli, Smith, Sheehan and Thompson20 For values of I 2GX <0.9, MR-Egger SIMEX corrections are presented using the highest of the two regression dilution statistics. For values of I 2GX <0.6, MR-Egger and MR-Egger SIMEX are unreliable and so neither are performed. Furthermore, mean F-statistics were calculated to assess the strength of the genetic instruments used.Reference Staiger and Stock25 As a rule of thumb, values of the mean F-statistic >10 indicate that the results do not suffer from substantially weak instrument bias.Reference Davies, Holmes and Davey Smith18 Lastly, we applied Steiger filtering to remove the genetic variants that explain more variance in the outcome than the exposure, to avoid possible reverse causation.Reference Hemani, Tilling and Davey Smith26 For smoking initiation, lifetime smoking and bipolar disorder, we reported the number of variants that were used in the bidirectional analyses after removing insertions or deletions and harmonisation of the data (Table 1).
Table 1 Bidirectional Mendelian randomisation (MR) analyses of the effects of smoking and bipolar disorder using summary-level data
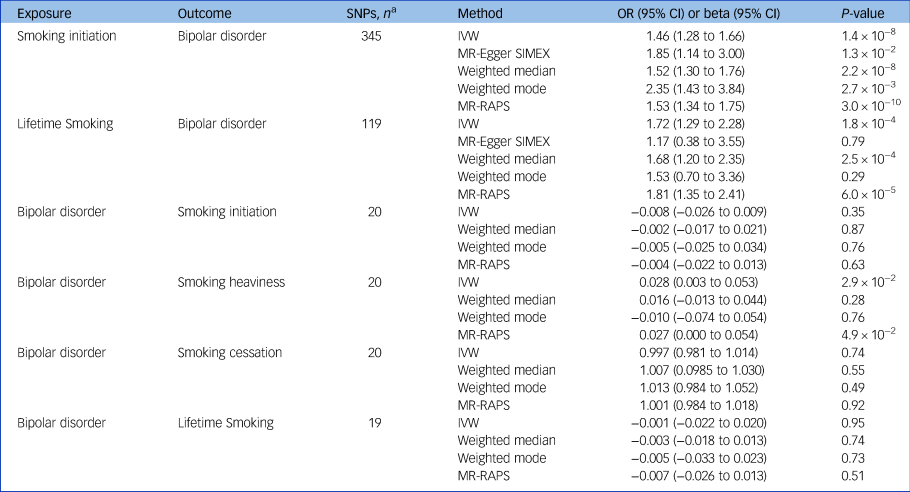
SNP, single nucleotide polymorphism; IVW, inverse-variance weighted; SIMEX, simulation extrapolation method; RAPS, robust adjusted profile score.
a. The number of SNPs used in each analysis after harmonisation of data-sets removing palindromic SNPs.
Results
Summary-level Mendelian randomisation of effect of smoking on bipolar disorder
Across different methods of MR analyses, consistent evidence was found for a positive effect of smoking initiation on the odds of bipolar disorder (ORIVW = 1.46, 95% CI 1.28–1.66, P = 1.4 × 10−8) (Table 1, Fig. 1, supplementary Figs 1–3). MR analyses using lifetime smoking as the exposure variable showed significant effects in the same direction, with a similar effect size (ORIVW = 1.72, 95% CI 1.29–2.28, P = 1.8 × 10−4) (Table 1, supplementary Figs 7–9). The I 2GX suggested using MR-Egger SIMEX results (Table 1, supplementary Table 1). Tests for heterogeneity suggest evidence for significant horizontal pleiotropy (supplementary Table 2), but MR-Egger intercepts suggest that bias of the estimates by pleiotropy is not likely (supplementary Table 3). To explore whether the genetic variants for lifetime smoking and smoking initiation explained more variance in the exposure than in the outcome, Steiger filtering was applied. For smoking initiation, 20 SNPS dropped out and 342/362 SNPS (94%) could be included in the analyses after Steiger filtering. For lifetime smoking, 12 SNPS dropped out and 107/119 SNPs (90%) could be included following Steiger filtering. The analyses were repeated across methods and similar effects were observed for smoking initiation and lifetime smoking on bipolar disorder (smoking initiation ORIVW = 1.44, 95% CI 1.29–1.62, P = 1.0 × 10−4; lifetime smoking ORIVW = 1.47, 95% CI 1.17–1.83, P = 7.9 × 10−4) (supplementary Table 5). Again, MR-Egger was not conducted as I 2GX values were below 0.9 (supplementary Table 1).
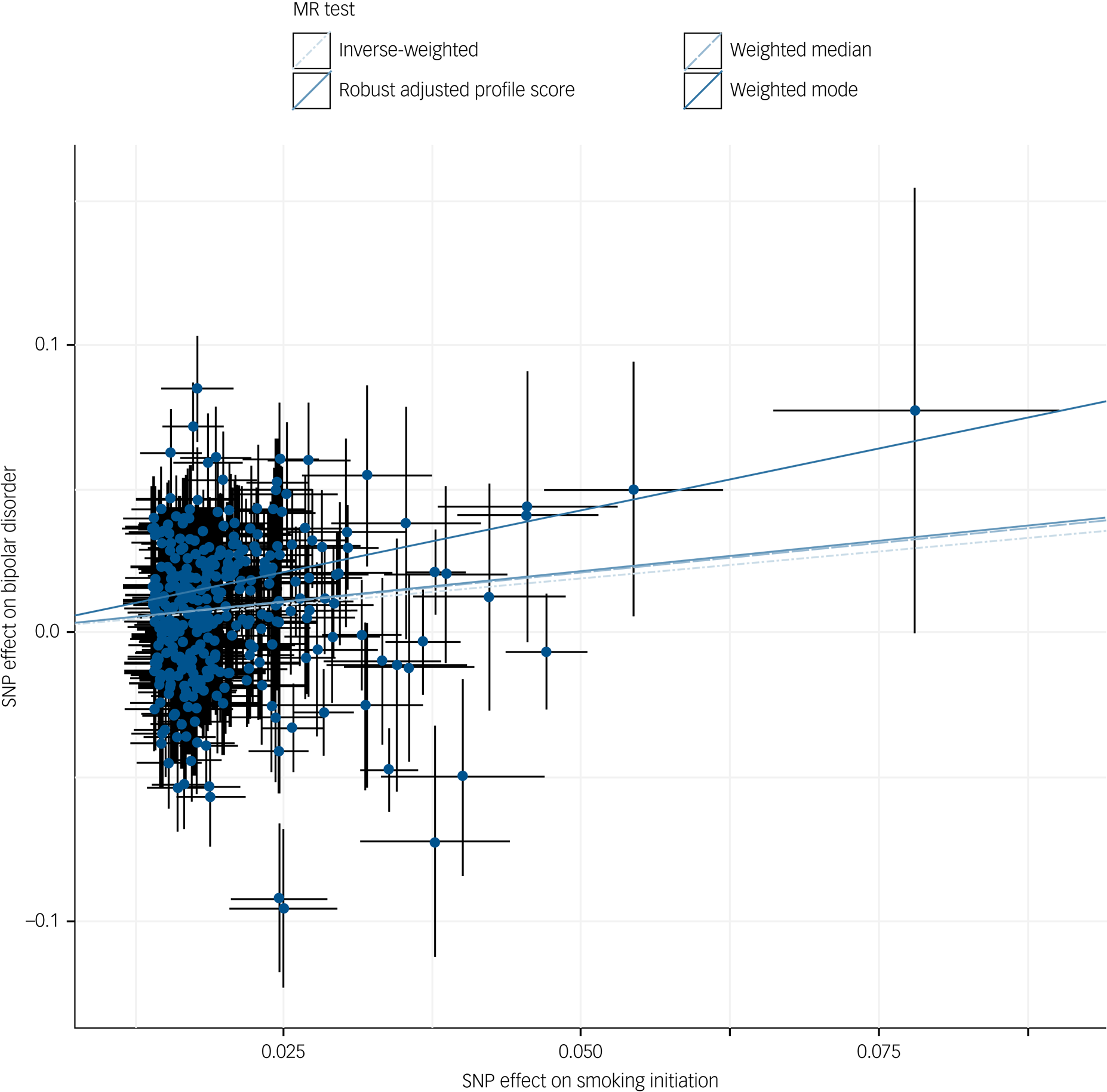
Fig. 1 Scatter plot of inverse-variance weighted and Mendelian randomisation (MR) Egger analyses of the effect of smoking initiation on bipolar disorder.
SNP, single nucleotide polymorphism.
Summary-level Mendelian randomisation of effect of bipolar disorder on smoking
The MR analyses of the effect of liability to bipolar disorder on risk of smoking initiation (betaIVW = −0.008, 95% CI −0.026 to 0.009, P = 0.35), lifetime smoking (betaIVW = −0.001, 95% CI −0.032 to 0.030, P = 0.95) and smoking cessation (ORIVW = −0.003, 95% CI 0.981–1.014, P = 0.74) revealed no evidence of a causal effect (Table 1, supplementary Figs 4–6, 10–12, 16–18). MR analyses of the effect of liability to bipolar disorder on smoking heaviness (betaIVW = 0.028, 95% CI 0.003–0.053, P = 2.9 × 10−2) showed significant small effects for IVW and MR-RAPS but not weighted median or weighted mode (Table 1, supplementary Figs 13–15). The I 2GX suggested not using MR-Egger or MR-Egger SIMEX results (supplementary Table 1). The tests for heterogeneity indicated no evidence for significant heterogeneity (supplementary Table 2) and MR-Egger intercepts suggested that bias of the estimates by horizontal pleiotropy is not likely (supplementary Table 3). Steiger filtering indicated that all genetic variants for bipolar disorder explained more variance in bipolar disorder than smoking outcomes. The mean F-statistic also suggested that results were not subject to weak instrument bias (supplementary Table 4). Although we did not find an effect using multiple genome-wide significant genetic variants, we noted that one SNP (rs3804640) stood out in particular, showing a large positive effect of bipolar disorder on smoking initiation and lifetime smoking in the single SNP analysis (supplementary Figs 5, 11). This SNP is an intron in the gene CD47, which encodes a membrane protein shown to play a role in synaptic plasticity in the hippocampus.Reference Lindberg, Gresham, Schwarz and Brown27–Reference Piccio, Vermi, Boles, Fuchs, Strader and Facchetti29
Discussion
The current study suggests that smoking is likely to be a causal risk factor for bipolar disorder by applying MR to two measures of smoking behaviour using GWAS summary statistics. In contrast, no evidence was found that liability to bipolar disorder has a causal effect on smoking initiation, smoking cessation or lifetime smoking. We found some evidence that suggested a small effect of liability for bipolar disorder on increased smoking heaviness. To the best of our knowledge, this is the first study providing robust evidence on the direction of the association between smoking and bipolar disorder.
Mendelian randomisation of effect of smoking on bipolar disorder
When comparing the current findings to those of a previous study evaluating the relationship between smoking and other severe mental illness,Reference Wootton, Richmond, Stuijfzand, Lawn, Sallis and Taylor12 the effect sizes we found are comparable to those testing lifetime smoking and smoking initiation as a risk factor for schizophrenia and major depressive disorder. The evidence for smoking as a risk factor for bipolar disorder has been meager.Reference Thomson, Berk, Dodd, Rapado-Castro, Quirk and Ellegaard7–Reference Slyepchenko, Brunoni, McIntyre, Quevedo and Carvalho9 However, one conventional population-based birth cohort study found that maternal smoking was a risk factor for bipolar disorder in offspring.Reference Talati, Bao, Kaufman, Shen, Schaefer and Brown6 To rule out the effect of maternal smoking as an explanation of our results, future cohorts with measures of maternal and offspring smoking with enough people with bipolar disorder are needed. In line with our findings, a study that used polygenic risk scores of smoking to predict bipolar disorder revealed a significant association but not vice versa.Reference Reginsson, Ingason, Euesden, Bjornsdottir, Olafsson and Sigurdsson30 Although this is suggestive of a causal effect, the current study adds to the body of evidence by overcoming residual confounding and providing an estimate of the causal effect size. The reported effect sizes of smoking in the current study are similar to those of other environmental risk factors (e.g. neurodevelopment, substances and physical/psychological stress) for developing bipolar disorder found in systematic reviews.Reference Marangoni, Hernandez and Faedda31,Reference Bortolato, Köhler, Evangelou, León-Caballero, Solmi and Stubbs32 Of note, those reviews included the following exposures as substance misuse: cocaine, cannabis, opioids, tranquillisers, maternal smoking during pregnancy. We argue that tobacco use needs to be considered for inclusion in this list.
Mendelian randomisation of effect of bipolar disorder on smoking
Although other studies have found an effect of bipolar disorder on smoking behaviour,Reference Heffner, Strawn, DelBello, Strakowski and Anthenelli8 the current study did not replicate this finding for most smoking behaviours. The lack of an effect indicates that liability to bipolar disorder is not a risk factor for smoking initiation, cessation or lifetime smoking. From a methodological point of view, one might explain the lack of a clear effect of the liability to bipolar disorder on smoking initiation, cessation and lifetime smoking by using weaker instruments for bipolar disorder as compared with smoking. This would result in reduced statistical power to detect a causal effect. However, we argue that this explanation is unlikely since the effect sizes consistently show values around the null, the confidence intervals were small and the mean F-statistic, which is a proxy for the strength of the instrumental variable, showed an acceptable value. The results for the effect of liability to bipolar disorder on smoking heaviness suggested from two MR methods a significant small effect and from two other methods no significant effect. This could indicate that liability to bipolar disorder increases smoking heaviness or there might be a bias from pleiotropy. Of note, MR typically investigates long-term effects of liability to bipolar disorder on lifetime smoking behaviour and firm conclusions on acute transient effects (e.g. more severe bipolar symptoms increasing the heaviness of smoking) could therefore not be drawn.
Strengths and limitations
A strength of this study is the substantial power, since we used summary data from recent large GWAS consortia to conduct a summary-level MR.Reference Burgess, Scott, Timpson, Smith, Thompson and Consortium33 Also, using genetic variants to determine the effects reduces the chance of reverse causation and residual confounding.Reference Lawlor, Harbord, Sterne, Timpson and Smith34 Furthermore, a consistent effect across different methods increases the confidence in most of our findings.Reference Lawlor, Tilling and Davey Smith24 Some limitations should be noted. First, the GWAS were conducted in individuals of European ancestry, which could limit generalisability to other populations. Second, although the smoking instruments capture smoking initiation, duration, heaviness and cessation, the instruments cannot be used to determine effects at specific life stages. The results should be interpreted as cumulative effects of smoking over the life course (potentially from conception and potentially from second-hand smoke from family members). In other words, it is not certain that our findings regarding smoking are confined to a specific period. Third, we assumed that the exclusion-restriction criteria hold since the MR-Egger intercept test for directional horizontal pleiotropy was not significant. Still, pleiotropy could be in place, as regression dilution values suggested that the MR-Egger results might not be trustworthy. The underlying biological process of mental illnesses are relatively poorly understood, which complicates the disentangling of plausible potential pathways. For example, genetic variants that were significantly associated with smoking initiation coded for several biologically plausible pathways, such as acetylcholine nicotinic receptors and glutamate pathways that affect reward processing and addiction.Reference Liu, Jiang, Wedow, Li, Brazel and Chen10 However, general biological pathways, such as sodium, potassium and calcium voltage-gated channels, were also implied, potentially overlapping with horizontal pleiotropic pathways. Nevertheless, results after Steiger filtering (e.g. testing whether the SNP explains more variance in the exposure or outcome and only selecting those explaining more in the outcome) showed similar effects of smoking on bipolar disorder. Although unlikely, pleiotropic effects could still be in place. Fourth, genetic vulnerability for smoking could also be related to environmental factors that are associated with the outcome, which could lead to spurious causal effects. Nevertheless, the consistent findings across all sensitivity analyses with different assumptions point towards robust findings. Unlike the prospective and longitudinal evidence concerning smoking and psychosis, epidemiological research regarding smoking as a risk factor for bipolar disorder is scarce. Future studies are needed to triangulate the evidence further. Fifth, sensitivity analyses into bipolar subtypes could not be performed because of the low number of associated genetic variants. Future GWAS with larger samples are required to potentially overcome this problem. Lastly, the bipolar disorder GWAS has a relatively low number of associated loci and a smaller sample compared with the smoking GWAS. Future GWASs with larger samples of people with bipolar disorder could yield a higher number of associated SNPs. An increase in the percentage of explained variance could alter MR results.
Possible mechanisms
There are several, non-mutually exclusive, plausible explanations for causal pathways from smoking to bipolar disorder.Reference Thomson, Berk, Dodd, Rapado-Castro, Quirk and Ellegaard7–Reference Slyepchenko, Brunoni, McIntyre, Quevedo and Carvalho9 First, nicotine binds to subtypes of the nicotinic acetylcholinergic receptor, which is implicated in the regulation of central nervous system pathways relevant to mood disorders.Reference Berk, Kapczinski, Andreazza, Dean, Giorlando and Maes35 Although the quality of the evidence is low, several case studies have related the use of a partial nicotine receptor agonist (varenicline) to induced mania or new onset of bipolar disorder.Reference Knibbs and Tsoi36–Reference Kohen and Kremen41 Second, nicotine exposure leads to desensitisation of the nicotine acetylcholinergic receptor by phospohorylation of subunits.Reference Mineur and Picciotto42 These dysmorphic receptors then have reduced binding capacity for endogenous acetylcholine: craving on drug withdrawal is clear evidence of changed function in reward pathways. The reduced functioning of these receptors could have an effect on neurotransmitter levels and subsequently lead to a higher vulnerability to develop bipolar disorder, although evidence about this topic is lacking. Third, burning tobacco releases toxic compounds that induce inflammation and oxidative stress, which may be associated with bipolar disorder.Reference Berk, Kapczinski, Andreazza, Dean, Giorlando and Maes35
Clinical implications
Many hypotheses exist to explain the high prevalence of smoking in severe mental illness.Reference Khokhar, Dwiel, Henricks, Doucette and Green43 The lack of a consistent bidirectional effect and the causal evidence that smoking initiation liability increases the risk for bipolar disorder point toward the two-hit or diathesis–stress model.Reference Fowles44 This implies a neurobiological vulnerability for severe mental illnessReference Lippard, Mazure, Johnston, Spencer, Weathers and Pittman45 interacting with an environmental stressor, such as smoking, leading to an increased vulnerability for severe mental illness.Reference Fowles44 Several studies have shown that smoking in adolescents and adults with bipolar disorder is associated with earlier age at onset of mood disorder, greater severity of symptoms,Reference Heffner, Anthenelli, Adler, Strakowski, Beavers and DelBello46–Reference Medeiros, Lafer, Kapczinski, Miranda-Scippa and Almeida51 poorer functioning, comorbid substance misuse,Reference Wilens, Biederman, Martelon, Zulauf, Anderson and Carrellas52 suicide attemptsReference Ducasse, Jaussent, Guillaume, Azorin, Bellivier and Belzeaux53 and other adverse outcomes, such as readmission to hospital.Reference O'Hagan, Cornelius, Young and Taylor54 Our results underline the importance of strategies to prevent adolescents from starting smoking and to treat smoking in people with bipolar disorder. The frequently mentioned reason for smoking by people with bipolar disorder is stress-coping behaviourReference Moon, Chang, Choi, Ha, Cha and Cho55 and a tailored prevention strategy might be helpful in learning healthy coping strategies. Regardless of the mechanism that may eventually be confirmed, our findings underline the importance of prevention and treatment programmes to reduce smoking and its consequences. Investing in a smoke-free future could reduce mental health problems in future generations.
Funding
R.E.W. is a member of the MRC Integrative Epidemiology Unit at the University of Bristol funded by the Medical Research Council (MC_UU_00011/7). This study was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre at the University Hospitals Bristol NHS Foundation Trust and the University of Bristol. The views expressed are those of the authors and not necessarily those of the National Health Service, the NIHR or the Department of Health.
Supplementary material
Supplementary material is available online at http://doi.org/10.1192/bjp.2019.202.





eLetters
No eLetters have been published for this article.