


Environmental challenge during development, including altered nutrition, is causally associated with an increased risk of cardio-metabolic disease in humans(Reference Gluckman, Hanson and Cooper1). Effects involve the transduction of signals about the prevailing environment from the mother to the fetus which act through developmental plasticity to alter the phenotype of the offspring. Several animal models show that suboptimal nutrition during critical periods in early development induces metabolic dysregulation that resembles human cardio-metabolic disease(Reference Gluckman, Hanson and Cooper1). In human subjects and in animal models, the nature of the phenotype induced by maternal dietary constraint differs according to the developmental stage of the fetus at the time of exposure(Reference Roseboom, de and Painter2, Reference Erhuma, Salter and Sculley3). Thus, the response of individual tissues to nutrient constraint is related to the stage of maturation, and hence the degree of plasticity, at the time of exposure.
Induction of altered phenotypes in response to environmental challenges such as nutrition during development involves changes in epigenetic processes, including methylation status of cytosines in CpG dinucleotides within the 5′-regulatory regions of genes and covalent modifications of histones(Reference Burdge and Lillycrop4). For example, feeding pregnant rats a protein-restricted diet induced hypomethylation and increased the expression of PPARα in the liver of the offspring, which was prevented by supplementation of the maternal protein-restricted diet with folic acid(Reference Lillycrop, Phillips and Jackson5), although some CpG in the PPARα promoter were hypermethylated compared to controls(Reference Lillycrop, Phillips and Torrens6), which implies targeting of the effects to individual CpG. Furthermore, maternal supplementation with methyl donors induced increased methylation and altered coat colour in Agouti viable yellow mice(Reference Wolff, Kodell and Moore7) and neonatal over-feeding induced hypermethylation of the pro-opiomelanocortin and insulin receptor promoters in the hypothalamus in mice(Reference Plagemann, Harder and Brunn8, Reference Plagemann, Roepke and Harder9).
For at least some genes, epigenetic plasticity may continue during the life course beyond early development(Reference Szyf10). For example, ageing is associated with a genome-wide decrease in DNA methylation(Reference Richardson11), T cell differentiation is dependent on reciprocal changes in the methylation of the IL-4 and interferon-γ promoters(Reference Sanders12), and carcinogenesis involves hypomethylation of cell-cycle genes and hypermethylation of tumour suppressor genes(Reference Burdge, Lillycrop and Jackson13). We have shown that dietary supplementation of rats with folic acid during their juvenile–pubertal phase induced a phenotype which superseded that induced by the maternal protein-restricted diet by inducing hypermethylation of the liver PPARα promoter and hypomethylation of the adipose tissue insulin receptor promoter which was associated with hepatosteatosis, dyslipidaemia and increased weight gain(Reference Burdge, Lillycrop and Phillips14). One potential implication of continuing plasticity is that it may be possible to modify differential disease risk induced during development by nutritional exposures later in the life course. However, the consequences of such interventions may be difficult to predict.
In order to understand better the effect of the timing of nutritional exposures on epigenetic regulation and phenotype, we compared the effect of supplementing pregnant rats with folic acid on glucose homeostasis in their offspring with the effect of supplementing the diets of juvenile rats with folic acid. Since maternal folic acid intake during pregnancy alters fasting glucose homeostasis in the offspring(Reference Burdge, Lillycrop and Jackson15), we used the epigenetic regulation of phosphoenolpyruvate carboxykinase (PEPCK) as a model system to test the effects of increased folic acid provision at different times in the life course. We measured the methylation status of individual CpG dinucleotides in the PEPCK promoter in the liver of male and female rats that had been fed a folic acid-adequate diet throughout life, or exposed to folic acid supplementation of their maternal or post-weaning diet.
Materials and methods
Animal procedures
The study was carried out in accordance with the Home Office Animals (Scientific Procedures) Act (1986) and approved by institutional ethical review. Virgin female Wistar rats (200–250 g) were fed RM1 chow diet (Special Diets Services) until conception when they were transferred to either a folic acid-adequate (AF) or an folic acid-supplemented (FS) diet until spontaneous delivery at about 21 d post-conception (Table 1). All bespoke and semi-purified diets were supplied by PMI Nutrition International and were based on the American Institute of Nutrition (AIN)-93 formulation with soyabean oil as the source of fat. Litters were standardised to eight (equal males and females) within 24 h of birth. Offspring were weaned on postnatal day 28 onto either an AF or an FS diet. Offspring were fed the FS diet for 28 d and then the AF diet. These regimens generated three groups of offspring for each sex (maternal diet:post-weaning diet): AF:AF (male n 9; female n 9), FS:AF (male n 5; female n 6) and AF:FS (male n 9; female n 9). Offspring were killed between postnatal days 84 and 90 after fasting for 12 h in order to standardise their metabolic state and to provide a nutritional challenge in order to assess their capacity to maintain glucose homeostasis. Livers were collected immediately into liquid N2 and stored at − 80°C. Blood was collected into tubes containing lithium heparin, centrifuged to separate cells from plasma and the plasma fraction stored at − 20°C. Plasma glucose concentration was measured by a standard automated colorimetric assay(Reference Burdge, Powell and Calder16). Plasma total folate concentration was measured by a standard colorimetric assay by the Department of Clinical Pathology, Southampton General Hospital, Southampton, UK.
Table 1 Composition of maternal and offspring diets
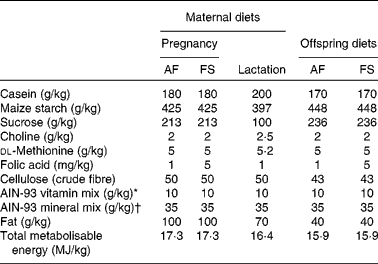
AF, folic acid adequate; FS, folic acid supplemented; AIN, American Institute of Nutrition.
* Vitamin mix (per kg mix): retinyl palmitate 13 μg; cholecalciferol 25 μg; dl-α-tocopherol acetate 83 mg; menadione 0·3 mg; thiamine hydrochloride 6 mg; riboflavin 6 mg; niacin 30 mg; calcium pantothenic acid 15 mg; pyridoxine hydrochloride 6 mg; biotin 0·2 mg; cyanocobalamin 25 μg.
† Mineral mix (per kg mix): Ca 5 g; available P 1·4 g; K 3·6 g; Mg 0·5 g; Na 1 g; Cl 2 g; F 10 mg; Fe 350 mg; Zn 340 mg; Mn 110 mg; Cu 60 mg; I 2 mg; Cr 10 mg; Mo 1 mg; Se 2 mg.
Measurement of phosphoenolpyruvate carboxykinase mRNA expression by real-time RT-PCR
Real-time RT-PCR was carried out essentially as described(Reference Burdge, Lillycrop and Phillips14). Briefly, total RNA was isolated from cells with TRIzol reagent according to the manufacturer's instructions (Invitrogen), and 1 μg was used as a template to prepare complementary DNA with 100 units of Moloney murine leukaemia virus RT. Complementary DNA was amplified using primers for PEPCK (forward AGCTGCATAATGGTCTGG, reverse GAACCTGGCGTTGAATGC) and for the housekeeping gene cyclophilin (forward TTGGGTCGCGTCTGCTTCGA, reverse GCCAGGACCTGTATGCTTCA). The reaction was performed in a total volume of 25 μl with SYBR Green Jumpstart Ready Mix (Sigma) as described by the manufacturer. Samples were analysed in duplicate, and C t values were normalised to cyclophilin(Reference Burdge, Lillycrop and Phillips14).
Measurement of phosphoenolpyruvate carboxykinase promoter methylation
The methylation status of individual CpG dinucleotides was measured in a region between 44 and 658 bp upstream from the PEPCK transcription start site(Reference Hoile, Lillycrop and Thomas17) which has known regulatory function(Reference Beale, Chrapkiewicz and Scoble18, Reference Yang, Reshef and Cassuto19) by pyrosequencing, essentially as described(Reference Lillycrop, Phillips and Torrens6). Briefly, genomic DNA was isolated(Reference Lillycrop, Phillips and Jackson5) and bisulphite conversion was carried out using the EZ DNA methylation kit (ZymoResearch). Modified DNA was amplified using hot startTaq DNA polymerase (Qiagen) by PCR using the following primers (locations are bp relative to the PEPCK transcription start site) − 658 to − 405 (forward AGGGGTTAGTATGTATATAGAGTGATT, reverse ATCAAAACACCACAACTATAAAATATC), − 417 to − 56 (forward GTGGTGTTTTGATAATTAGTAGTGATT, reverse CCCCTCAACTAAACCTAAAAACTC) and − 373 to − 44 (forward GTTAGTAGTATATGAAGTTTAAGA, reverse CCCCTATTAACCAAAAATATATTCC). PCR products were immobilised on streptavidin–sepharose beads (Amersham), washed, denatured and released into annealing buffer containing the following sequencing primers: GTGATTATTTTATATTAGGTATTG, AGAGGATTTAGTAGATATTTAGTG, TAAATATTAAAAAACCTCAAACCC, GGTTAAAGTTTAGTTAATT and TTATTATTTTTTTAAAGTTTATTG. Pyrosequencing was carried out using the SQA kit on a PSQ 96MA machine (Biotage) and percentage methylation was calculated using the Pyro Q CpG programme (Biotage). Within-assay precision was between 0·8 and 1·8 %, and between-assay variation was between 1 and 3 % depending on CpG. Detection limits were 2–5 % methylation. Between-assay CV were CpG − 606, 3 %; CpG − 508, 1·4 %; CpG − 440, 2·2 %; CpG − 248, 1·7 %; CpG − 129, 1·7 %; CpG − 100, 1·9 %; CpG − 90, 2·0 %; CpG − 81, 1·0 %.
Electromobility shift assay
Electromobility shift assay was used to determine the effect of methylation of CpG − 248 on protein binding at the locus(Reference Harris, White and Phillips20). Oligonucleotides containing unmethylated or methylated CpG − 248 (5′-AGTCAATCAAACGTTGTGTAAGGACTCAACTA-3′ and 3′-TAGTTGAGTCCTTACACAACGTTTGATTGACT-5′) were synthesised by Biomers. Nuclear extracts were prepared as described(Reference Dignam, Lebovitz and Roeder21). Electromobility shift assay was carried out using the LightShift Chemiluminescent Electro Mobility Shift Assay Kit (Thermo Scientific) as instructed by the manufacturer. Competitions were performed using 100-fold excess of unlabelled oligonucleotide, which was incubated with the nuclear extracts before the addition of the probe. The effect of methylation of CpG − 248 on the binding of CCAAT-enhancer-binding protein (C/EBPβ) was determined using a polyclonal antibody (Abcam ab32358) as described(Reference Harris, White and Phillips20).
Statistical analysis
Values are represented as mean (1 sd). Comparisons between groups were by a general linear model with maternal diet, postnatal diet and sex as fixed factors, with Bonferroni's post hoc test. The results of real-time RT-PCR analysis were found to be non-normally distributed and so were log10 transformed before analysis. Analysis of the relationship between PEPCK CpG methylation and mRNA expression was by bivariate correlation using Pearson's test.
Results
There were no significant differences between groups in food intake, body weight or liver gross morphology (data not shown). There were no significant differences in plasma folate status between groups of adult offspring (male AF:AF, 22 (sd 4) μg/l; FS:AF 20 (sd 4) μg/l; AF:FS, 20 (sd 3) μg/l; female AF:AF, 21 (sd 4) μg/l; FS:AF, 20 (sd 2) μg/l; AF:FS, 20 (sd 4) μg/l). There were significant interactive effects of sex × maternal diet (P = 0·002) and sex × post-weaning diet (P = 0·003) on plasma glucose concentration. There were no significant differences between groups of male offspring, while plasma glucose was significantly higher in FS:AF females and lower in AF:FS females compared to AF:AF females and to all groups of males (Fig. 1). There were significant interactive effects of sex × maternal diet (P = 0·041) and sex × post-weaning diet (P = 0·02) on hepatic PEPCK mRNA expression. The pattern of PEPCK expression was similar to that of plasma glucose (Fig. 1). There was no significant association between fasting plasma glucose concentration and PEPCK mRNA expression in male offspring, while PEPCK mRNA expression was significantly positively correlated (r 0·37, P = 0·003) with plasma glucose concentration in females.

Fig. 1 Plasma glucose concentration, phosphoenolpyruvate carboxykinase (PEPCK) mRNA expression and promoter methylation for AF:AF (□) (male n 9; female n 9), FS:AF (■) (male n 5; female n 6) and AF:FS (![]() ) (male n 9; female n 9) offspring. CpG dinucleotides are indicated by the position (in bp) relative to the transcription start site. Values are means, with standard deviations represented by vertical bars. Statistical comparisons were by a general linear model with Bonferroni's post hoc test. a,b,c Mean values within a sex with different letters were significantly different (P < 0·05). * Mean value was significantly different from that of the male rats of the same dietary group (P < 0·05). AF, adequate folic acid; FS, folic acid supplemented. Groups were (maternal diet:post-weaning diet): AF:AF, FS:AF and AF:FS.
) (male n 9; female n 9) offspring. CpG dinucleotides are indicated by the position (in bp) relative to the transcription start site. Values are means, with standard deviations represented by vertical bars. Statistical comparisons were by a general linear model with Bonferroni's post hoc test. a,b,c Mean values within a sex with different letters were significantly different (P < 0·05). * Mean value was significantly different from that of the male rats of the same dietary group (P < 0·05). AF, adequate folic acid; FS, folic acid supplemented. Groups were (maternal diet:post-weaning diet): AF:AF, FS:AF and AF:FS.
Increasing the folic acid content of the maternal or post-weaning diets induced changes in the methylation of specific CpG dinucleotides in the PEPCK promoter in female, but not male, offspring (Fig. 1). There was a significant effect of sex (P < 0·0001), post-weaning diet (P < 0·0001) and sex × post-weaning diet (P = 0·002) on the methylation of CpG − 606, of sex (P = 0·025), maternal diet (P < 0·0001) and sex × maternal diet (P = 0·003) on CpG − 508, of maternal diet (P = 0·02) and post-weaning diet (P = 0·002) on CpG − 440, of post-weaning diet on CpG − 248 (P = 0·032), CpG − 129 (P = 0·007) and CpG − 90 (P = 0·036), and of post-weaning diet (P = 0·005), sex × maternal diet (P = 0·01) and sex × post-weaning diet (P < 0·0001) in CpG − 81. There was no significant association between CpG methylation and PEPCK mRNA expression or plasma glucose concentration in males. However, in females, PEPCK mRNA expression and plasma glucose concentration were correlated with the methylation level of CpG − 606 (r 0·417, P = 0·001; r 0·702, P < 0·0001), CpG − 440 (r − 0·533, P = 0·009; r − 0·427, P = 0·033), CpG − 248 (r − 0·561, P = 0·011) and CpG − 81 (r − 0·412, P = 0·006; r − 0·412, P = 0·041).
Methylation of CpG − 248 decreased specific protein binding (Fig. 2). Incubation with an antibody against C/EBP decreased the mobility of the methylated oligonucleotide–protein complex but did not alter the mobility of the unmethylated probe.

Fig. 2 Analysis of protein binding at CpG − 248 by electromobility shift assay. Lanes are: (1) unmethylated probe, (2) unmethylated biotinylated probe plus nuclear extract, (3) unmethylated biotinylated probe plus nuclear extract plus unbiotinylated probe; indicates specific protein binding, (4) unmethylated probe plus nuclear extract plus non-specific biotinylated probe; confirms specific protein binding, (5) unmethylated probe plus nuclear extract plus anti-CCAAT-enhancer-binding protein β (C/EBPβ); indicates C/EBPβ binding, (6) methylated probe, (7) methylated biotinylated probe plus nuclear extract, (8) methylated biotinylated probe plus nuclear extract plus unbiotinylated probe, (9) methylated probe plus nuclear extract plus non-specific biotinylated probe, (10) methylated probe plus nuclear extract plus anti-C/EBPβ. Data are representative of three experiments. (A) Migration of the anti-C/EBPβ antibody–oligonucleotide–nuclear extract complex. (B) Migration of the oligonucleotide–nuclear extract complex.
Discussion
The findings of the present study show that the epigenetic regulation of PEPCK differed between the offspring of dams fed an FS diet and those fed an FS diet after weaning in a manner contingent on sex.
Liver glycogen is almost completely depleted in rats fasted for 12 h(Reference Freedland22), and hence plasma glucose homeostasis depends primarily on the balance between hepatic gluconeogenesis and glucose uptake. Folic acid supplementation altered glucose homeostasis and the epigenetic regulation of PEPCK in female, but not male offspring. Such sex differences are consistent with previous findings which show differential effects of maternal dietary exposures on the phenotype of male and female offspring(Reference Gilbert and Nijland23) and with general observations of susceptibility to non-communicable diseases between males and females(Reference Kautzky-Willer and Handisurya24). The absence of significant differences in folate status between groups suggests that any phenotypic differences induced by the different diets were not due to induced changes in folate metabolism. The mechanism underlying such sex-dependent changes is not known, but implies an interaction between the mechanisms which regulate epigenetic marks and sex hormones, even in immature tissues. In females, the route and/or timing of increased folic acid exposure determined the nature of the effect on glucose concentration and PEPCK mRNA expression. Both maternal and post-weaning folic acid supplementation decreased the capacity to maintain glucose homeostasis, although the effects were in opposite directions between time points. This implies that, at least for folic acid, the same dietary exposure cannot be assumed to induce the same effects on phenotype at all stages of the life course. However, although the same amount of folic acid was provided in the maternal and post-weaning diets, it cannot be assumed that, because of maternal and placental folate metabolism, the fetus and juvenile offspring were exposed to the same increase in folate exposure. Thus, at least part of the difference in the effect of maternal and post-weaning folic acid supplementation on the epigenome and phenotype of the offspring may have been due to differences in the actual level as well as the timing of increased folic acid supplementation.
Folic acid supplementation has been shown to induce gene-specific and global changes in regenerating axons(Reference Iskandar, Rizk and Meier25), to alter coat colour in Agouti mice(Reference Wolff, Kodell and Moore7), to induce persistent changes in liver global methylation(Reference Kotsopoulos, Sohn and Kim26) and to increase H19 methylation in patients with ureamia(Reference Ingrosso, Cimmino and Perna27). Maternal, but not post-weaning, folic acid supplementation in rats reduced global DNA methylation in non-neoplastic mammary tissue(Reference Ly, Lee and Chen28). Furthermore, the methylation profile of individual CpG in human cord blood has been shown to be associated with homocysteine status(Reference Fryer, Emes and Ismail29) which implies differential vulnerability of individual CpG to variations in capacity for 1-carbon metabolism in the epigenome of the developing human fetus. Our findings show for the first time the specific effects of folic acid supplementation on the methylation of individual CpG in offspring which were exposed to increased folic acid via the mother or after weaning. For example, CpG − 508 was only altered in female offspring exposed to folic acid supplementation before birth, CpG − 606 and − 248 were only altered in offspring which received the supplemented diet after weaning, while CpG − 440, − 129 and − 81 were altered by folic acid supplementation irrespective of the timing of the exposure. Such specificity may reflect the differential activation of DNA methyltransferases in response to the folic acid supplement(Reference Ly, Lee and Chen28, Reference Ghoshal, Li and Datta30) together with the targeting of DNA methyltransferases to individual CpG(Reference Robertson, it-Si-Ali and Yokochi31).
In females, both PEPCK mRNA expression and the methylation of CpG − 606, − 440, − 248 and − 81 were related statistically to fasting plasma glucose concentration. This suggests that the epigenetic regulation of PEPCK is an important factor for maintaining glucose homeostasis in rats, although a direct causal association between PEPCK methylation and expression requires further experimentation. The association between methylation of CpG − 606 and PEPCK expression and glucose homeostasis was positive, while the associations with CpG − 440, − 248 and CpG − 81 were negative. While CpG − 606 is not located within a predicted transcription factor binding site, CpG − 440 is within a PPAR response element, CpG − 248 is within a CCAAT-enhancer response element and CpG − 81 is within a cyclic AMP response element(Reference Hoile, Lillycrop and Thomas17–Reference Yang, Reshef and Cassuto19), both of which regulate PEPCK transcription(Reference Srivastava32, Reference Thonpho, Sereeruk and Rojvirat33). Thus, differences in the nature of the association between the methylation status of individual CpG and the level of transcription may reflect the interaction between specific regulatory proteins and the DNA sequence. Methylation of CpG − 248 decreased overall binding at this locus, but increased specific binding of C/EBPβ. This suggests that methylation of CpG − 248 may increase the transcriptional enhancer activity of C/EBPβ. If so, hypomethylation at this locus in female offspring fed the FS diet after weaning may explain, at least in part, the lower PEPCK mRNA expression in this group. Binding of C/EBPβ to its response element is inhibited by binding of GATA-1 in the presence of transcriptionally active histone modifications(Reference Burda, Curik and Kokavec34). Thus, one possible mechanism by which methylation of CpG − 248 may increase C/EBPβ binding is through displacement of a negative regulator.
Overall, the findings of this study suggest that the timing and/or route of supplementation with folic acid induced sex-related, differential effects on the epigenetic regulation of PEPCK and on glucose homeostasis. Irrespective of the underlying mechanism, these findings demonstrate that folic acid supplementation at different times in development can alter the epigenotype and phenotype, specifically the capacity to maintain fasting glucose homeostasis, in the offspring which is sustained into adulthood. Overall, these findings are consistent with a number of experimental studies which show that folic acid supplementation during development and later in the life course can induce long-term changes in gene regulation and expression(Reference Lillycrop, Phillips and Torrens6, Reference Wolff, Kodell and Moore7, Reference Burdge, Lillycrop and Phillips14, Reference Ly, Lee and Chen28, Reference Waterland, Dolinoy and Lin35–Reference Caldwell, Manziello and Howard37). If these findings were replicated in human subjects, they would have important implications for recommendations on folic acid supplementation and fortification, and would suggest an urgent need for further investigation and re-evaluation of the safety of folic acid.
Acknowledgements
The present study was funded in part by a Fellowship grant from the British Heart Foundation (FS/05/064/19525) to G. C. B. and by departmental funds. S. P. H. receives salary support from the BBSRC and M. A. H. from the British Heart Foundation (CH/02/01). G. C. B., S. P. H. and K. A. L. designed the study; S. P. H. and L. R. G. performed the experiments; G. C. B. analysed the data and wrote the paper with K. A. L. and M. A. H. The authors declare that there are no conflicts of interest. The authors thank Dr E. S. Garratt for assistance with the analysis of gene expression.



