



The polysaccharide capsule of Klebsiella pneumoniae is regarded as a virulence factor, protecting it against the host immune response. Seventy-nine capsular types have been identified [Reference Pan1–Reference Hsu3]. Some of these capsular types have been associated with invasive disease, largely because of particular ‘hypervirulent’ strains belonging to them, most notably clonal groups (CG) 23 (of capsular type K1), 86, 375 and 380 (capsular type K2) all of which have been strongly associated with sepsis and liver abscesses, often in otherwise healthy people [Reference Bialek-Davenet4, Reference Holt5]. Compared with those belonging to capsular types K1 and K2, isolates belonging to other capsular types have been relatively little studied. Nevertheless, capsular type K54 has also been associated with hypervirulence, particularly representatives of sequence type (ST) 29 [Reference Iwasaki6–Reference Hsu10]; the presence of a large virulence plasmid carrying capsule-upregulation genes rmpA/rmpA2 and the aerobactin siderophore cluster is also important. We sought to investigate K. pneumoniae submitted to the UK national reference laboratory and positive for the K54 capsular type for evidence of hypervirulence. In doing so, it quickly became clear that one type (with variants), corresponding to CG29, dominated and that we had both K54 and non-K54 representatives of it, the latter including the reference strain NCTC 9159 [11], the whole-genome sequence of which had been determined in the NCTC 3000 project [12]. In addition, whole-genome sequences were available of two isolates of capsular type K54 and of ST29; one from liver abscess puncture fluid from China (LS358, CP025629) and the other from the environment in Australia (KP-1, CP012883). This provided a rare opportunity to be able to compare these isolate sets, with the aim of providing potential insights into the hypervirulent phenotype and the importance or otherwise of CG and capsular type.
Bacterial isolates
Between 2012 and the end of June 2017, 5566 isolates of K. pneumoniae, including those from 4670 patients, were typed by the laboratory, of which 42 were of capsular type K54 and/or of variable number tandem repeat (VNTR) type 7,3,1/7,5,1,2,3/4,1,2/3/4,2,4/5 (corresponding to CG29) and were included in this study. Clinical details were obtained from information from the request form completed by the sending hospital accompanying each isolate.
Typing and PCR detection of capsular types and virulence genes
Isolates were typed by VNTR analysis at 11 loci (A, E, H, J, K, D, N1, N2, 52, 45, 60 in that order) as described previously [Reference Turton13]; those no longer in our archives had been typed at nine loci, of which the first eight were in common with those in the 11-locus scheme. Detection of capsular types K1, K2, K5, K20, K54 and K57 and of rmpA, rmpA2 and wcaG was by multiplex PCR [Reference Turton13, Reference Turton14]. Virulence genes iutA, allS, rmpA, mrkD and ybtS were sought by a further multiplex PCR using the primers and conditions of Compain et al. [Reference Compain15], together with iroD (using primers iroDF, 5′-GTATGCGCGGCAATTGAC-3′ and iroDR, 5′-GAACGGTCTGCCCACTG) and clbR (using primers clbRF, 5′-CATGCTGGCACAAGGTGA and clbRR, 5′-CGTGATTCGTATTCCGAGC-3′) giving products of 920, 764, 461, 340, 242, 194 and 122 bp, respectively. The iroD and cblR primers were designed taking into account all the alleles of these genes available from the multilocus sequence typing (MLST) ‘download allele sequences’ page [16] and were present at a final concentration of 0.1 pmol/μl. These additional targets did not affect the performance of the PCR, which gave the same results for the iutA, allS, rmpA, mrkD and ybtS targets as the 5-plex alone. The 7-plex PCR was validated against a panel of 14 isolates carrying various combinations of these genes, and which had previously undergone whole-genome sequencing and detection of virulence genes from the sequences. DNA extracted from isolate KpvST23_OXA-48, which was positive for all seven targets, was used as positive control [Reference Turton13]. MLST was carried out as described by Diancourt et al. [Reference Diancourt17].
Nanopore sequencing of isolate KpvK54
Genomic DNA was extracted from KpvK54 using a GeneJet kit (ThermoFisher, Loughborough, UK). The whole-genome sequence was determined on an Mk1B minION using a MIN107 flow cell and an SQK-LSK308 1D2 sequencing kit (R9.5 chemistry) (Oxford Nanopore technologies). Library preparation was carried out following the protocol described on the nanoporetech community pages with no additional shearing step. Briefly, 1 µg genomic DNA was end-repaired/dA tailed using New England Biolabs (NEB) Ultra II End-prep reaction buffer/enzyme mix (E7546), ligated with 1D2 adapter using NEB Blunt/TA Ligase Mix (M0367) and adapter ligated using Ampure XP beads to purify the DNA between each step. DNA was eluted in a volume of 15 µl to give the pre-sequencing mix, which was then mixed with running buffer with fuel mix and Library Loading Bead kit (Oxford Nanopore technologies) and loaded onto a primed flow cell. Sequencing was carried out using the NC_48Hr_Sequencing_Run_FLOMIN107_SQK-LSK308 script on minKNOW (version 1.2.8).
Reads were basecalled using Albacore (ont-albacore-1.1.2-amd64) and assembled with miniasm 0.2-R159-dirty and then corrected with three iterations of Racon [Reference Vaser18], using minimap 0.2-R124-dirty to map the reads to the assembly [Reference Li19]. Following the initial analysis, a new version of Albacore (ont-albacore-2.0.2-amd64) was released and the basecalling was repeated; this analysis gave 1D2 pass reads, which were used to correct the previous analysis, again with three iterations of Racon. Additionally, assembly using Canu (version 1.5 +102 changes) was performed [Reference Koren20]. The sequences were deposited in GenBank under accession numbers CP023134–CP023137. The original albacore basecalled reads fastq file, together with the 1D2 pass reads, is available under STUDY: PRJNA400335 (SRP118927), SAMPLE: KpvK54 (SRS2542913), EXPERIMENT: KpvK54_minION (SRX3218008) RUN: K54.fastq.gz (SRR6078568) and can be accessed at https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP118927.
Whole-genome sequence of NCTC 9159
The whole-genome sequence of NCTC 9159 was obtained from http://www.sanger.ac.uk/resources/downloads/bacteria/nctc/ (ftp://ftp.sanger.ac.uk/pub/project/pathogens/NCTC3000/datalinks_manual/ERS686710.gff). Virulence genes and wzc and wzi alleles downloaded from the bigsdbMLST database [16] or from NCBI nucleotide were detected from whole-genome sequences using BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch).
Identification of integrative conjugative element (ICEKp) in KpvK54
The ICEKp was identified by comparing against the 14 sequences available on ‘Kleborate’ [21] (in Kleborate\build\lib.linux-x86_64-2.7\kleborate\ICEKp-references).
Comparison of gene contents of KpvK54, NCTC 9159, LS358, KP-1, NTUH-K2044 and RJF999
The coding sequences from the chromosome sequence of KpvK54 (utg000001c, CP023134) were identified using Prodigal version 2.6.1 [Reference Hyatt22]. These, together with the annotated coding sequences from the chromosome sequences of NCTC 9159, LS358 (CP025629), KP-1 (CP012883), NTUH-K2044 (AP006725) and RJF999 (CP014010), were put through cd-hit version 4.7 [Reference Fu23] to cluster similar genes together using the default setting of 90% similarity. To ensure that genes were not missed in any of the genomes, a representative sequence from each cluster was compared against the entire chromosome sequences using a local BLAST database (BLAST 2.2.29+) and any matches added to the relevant cluster. A matrix of presence or absence for each coding sequence in the six genomes was made and genes put into sets depending on which combinations of the genomes in which they were found.
Six genes identified as potentially associated with virulence from this approach were further sought by PCR in a subset of isolates representing all three groups described in Table 1, using the primers detailed in Supplementary Table S1. In addition, K54 isolates of other types were included. PCRs were carried out using the QIAGEN Taq core kit with denaturation at 95 °C for 3 min, followed by 35 cycles of 95 °C for 30 s, 60 °C for 30 s, 72 °C for 1 min, followed by a final extension at 72 °C for 10 min.
Table 1. Isolates of VNTR type 7,3,1/7,5,1,2,3/4,1,2/3/4,2,4/5 received from UK hospitals between 2012 and end of June 2017

a Isolates were labelled by patient (P1–25), hospital, whether K54-positive, or K54-positive with evidence of virulence plasmid (K54v) and date received (week.year).
b Hospitals were labelled according to the region and number within that region; L = London, NE = North East England, NW = North West England, SE = South East England, M = Midlands.
c Isolates no longer in our archives and typed only with a nine-locus VNTR scheme, of which the first eight loci are in common with the 11-locus scheme used for the remainder.
d nt; not tested.
Results
We had recorded 31 non-duplicate isolates belonging to capsular type K54 on our database, of which the largest group (15) were of the types 7,3,1/7,5,1,2,3/4,1,2/3/4,2,4/5, with most (⩾9) having the profile 7,3,1,5,1,2,4,1,3/4,2,4; there was clear evidence for hypervirulence among isolates of this type (blood isolates, rmpA/rmpA2-positive, clinical details of septicaemia, abscesses, query of hypervirulence from requesting hospitals) (Table 1A) (Fig. 1). All of these were also PCR-positive for wcaG, always found in K54 isolates. We also had 11 K54-negative (and wcaG-negative) isolates of this type on our database (Table 1C), with many of these associated with carbapenemase genes; none were PCR-positive for rmpA/rmpA2 or iutA, indicating lack of a virulence plasmid; only three of the 11 isolates were from blood, but they also included those from pus (from an abscess) and pleural fluid. One of these was a reference isolate, NCTC 9159 [11], of capsular type 39 and ST985 (a single locus variant (SLV) of ST29), isolated from urine in 1952 in Denmark, the whole-genome sequence of which had been determined in the NCTC 3000 project [12] (shown in green in Fig. 1).
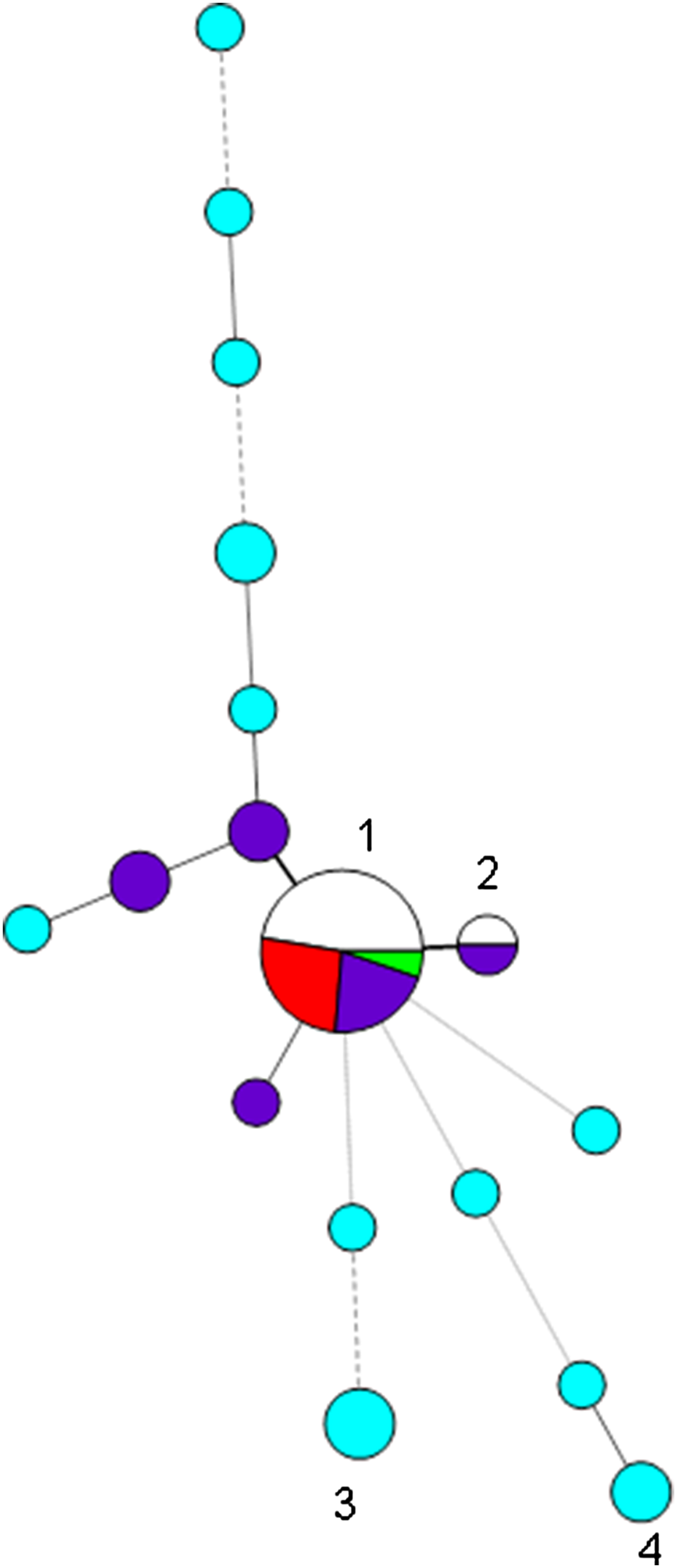
Fig. 1. Minimum spanning tree of variable number tandem repeat (VNTR) profiles at the first eight loci of the 42 isolates belonging to capsular type K54 and/or of profile 7,3,1/7,5,1,2,3/4,1,2/3/4,2,4/5 (corresponding to CG29) found during 2012–2017. Each circle represents a type and the size of the circle is proportional to the number of isolates of that type. Circles 1 and 2 denote profiles beginning 7,3,1,5,1,2,4,1 (19 isolates) and 7,3,1,-,1,2,4,1 (two isolates), respectively; segments marked in red denote K54 isolates of CG29-positive for rmpA/rmpA2, those in dark blue K54 isolates attributed to CG29 in which rmpA/rmpA2 were not detected, and uncoloured/green segments those of CG29-negative for K54 and negative for rmpA/rmpA2; NCTC 9159 is denoted by the green segment. K54 isolates not of CG29 are denoted by turquoise circles, e.g. circles 3 and 4 denote isolates with profiles beginning 4,1,3,2,2,2,2,3 and 1,-,2,4,0,1,2,5. Thick solid lines join single-locus variants, while thinner, dashed or dotted lines join multilocus variants (up to three, four and six loci different, respectively).
These 26 isolates (15 K54-positive and 11 K54-negative) sharing similar profiles (7,3,1/7,5,1,2,3/4,1,2/3/4,2,4/5) could therefore be divided into three groups: (i) K54-positive isolates that were PCR-positive for rmpA, rmpA2, iutA and iroD, indicating the presence of a virulence plasmid carrying both the aerobactin and salmochelin siderophore clusters, and for ybtS from the yersiniabactin siderophore cluster (5); (ii) K54-positive isolates that were PCR-negative for rmpA, rmpA2, iutA and iroD and clbR, indicating lack of a virulence plasmid, with ybtS variably present (10) (Table 1B); and (iii) K54-negative isolates lacking evidence of a virulence plasmid, with ybtS variably present (11). Isolates with the virulence plasmid had 2 or 3 repeats at the ninth VNTR locus, where determined, while the other isolates mostly had a repeat of 4 or 5 at that locus (16/17). The non-K54 representatives were more likely to have a carbapenemase gene (6/11) than the K54 examples (2/15) (P = 0.04 by two-tailed Fisher's exact test); three of these isolates were from rectal/faecal screens. We did not detect clbR (from the colibactin cluster) in any of the isolates.
The whole-genome sequence of isolate KpvK54, which belonged to the first group (i; group A in Table 1) and was isolated in 2017 from pus, was determined by nanopore sequencing. The sequence was assembled from 363 659 reads (total 4 176 296 833 bases) with an average read length of 11 484 bases and 4156 reads longer than 46 879 bases; the longest read was 122 975 bases. The whole-genome sequence confirmed the capsular type of K54 (wzc_54). The sequence obtained was at very high coverage (783-fold), and therefore provided complete assemblies, but was not entirely accurate (99% according to BLAST searches and comparison with Sanger sequencing of the MLST housekeeping genes) mainly because of systematic problems with homopolymer runs (runs of the same base), which are a recognised problem with minION sequences. Correction of the initial analysis with the 1D2 pass reads basecalled with the later version of Albacore improved the accuracy. The final miniasm/Racon-corrected assembly gave four contigs of 5 224 722 bp (utg000001c, the chromosome, CP023134), 211 454 bp (utg000002c, the virulence plasmid, CP023135), 31 074 bp (utg000003 l, CP023136), which has 79% homology with an extrachromosomal sequence described in K. pneumoniae CAV1217 (CP018673) and 12 361 bp (utg000004c, CP023137), which matched parts of a plasmid found in Escherichia coli (pEC15II, KU932031.1 and pDKX1-TEM-52, JQ269336). The Canu assembly (from the original basecalled reads) gave two contigs of length 5 230 099 bp (tig0000001) and 263 869 bp, neither of which was circular; we chose to use the miniasm/Racon-corrected assembly. Sanger sequencing confirmed the MLST type (ST2849) as a double locus variant of ST29 of allelic profile 158,3,2,2,6,4,160 (with alleles differing from ST29 underlined) and a SLV of NCTC 9159. The most similar isolate on the MLST database (isolate id 769) belongs to ST714, a SLV from a patient from Asia with bacteraemia [24].
KpvK54 had a large virulence plasmid (211 kb) carrying the aerobactin (iutA, iucABCD) and salmochelin (iroBCDN) acquired siderophore systems as well as rmpA and rmpA2 and lead (pbrABCR), copper (pcoABCDERS), silver (silCERS) and tellurium (terABCDEWXYZ) resistance genes, typical of hypervirulent types (Supplementary Table S2). The yersiniabactin cluster (ybtAEPQSTUX and fyuA, irp1, irp2) was also present on the chromosome, so that the isolate carried three acquired siderophore systems, strongly correlated with hypervirulence. It also contained other virulence genes sought, both on the plasmid (hemin, SAM-dependent methyltransferase, luxR, pagO, ibrB, fecA, fecI, fecR, Fe 3+ citrate ABC transporter, fur, shiF, lysozyme inhibitor, cobW) and on the chromosome (mrkABCDFHIJ (type 3 fimbrial genes)).
In common with most hypervirulent isolates, which are typically susceptible to most antibiotics, isolate KpvK54 contained few resistance genes: with bla SHV-83 (penicillinase non-extended spectrum β-lactamase), oqxA and oqxB (fluoroquinolone) (core genes in K. pneumoniae) and fosA (fosfomycin) detected on the chromosome.
The NCTC 9159 chromosome sequence (called ‘unitig_0_quiver|quiver’) covered 96% of that of KpvK54 with 99% identity (BLAST total score of 9.984 × 106), highlighting the similarity between these two isolates, despite isolation 65 years apart (Fig. 2). On BLAST analysis, the KpvK54 chromosome was also closely similar to those of LS358 (CP025629) (98% coverage and 99% identity, BLAST total score 9.627 × 106) and KP-1 (CP012883) (96% coverage and 99% identity, BLAST total score of 1.006 × 107); both isolates are also of capsular type K54 and belong to ST29 with LS358 having been isolated from liver abscess puncture fluid, while KP-1 was from the environment. Other best matches appearing on BLAST comparisons were with NTUH-K2044 (AP006725) (total score of 9.482 × 106) and RJF999 (CP014010) (total score of 9.316 × 106) which had 92% and 93% coverage, respectively, and 98% identity with the KpvK54 chromosome. Both NTUH-K2044 and RJF999 belong to K1-ST23, the type most associated with hypervirulence.
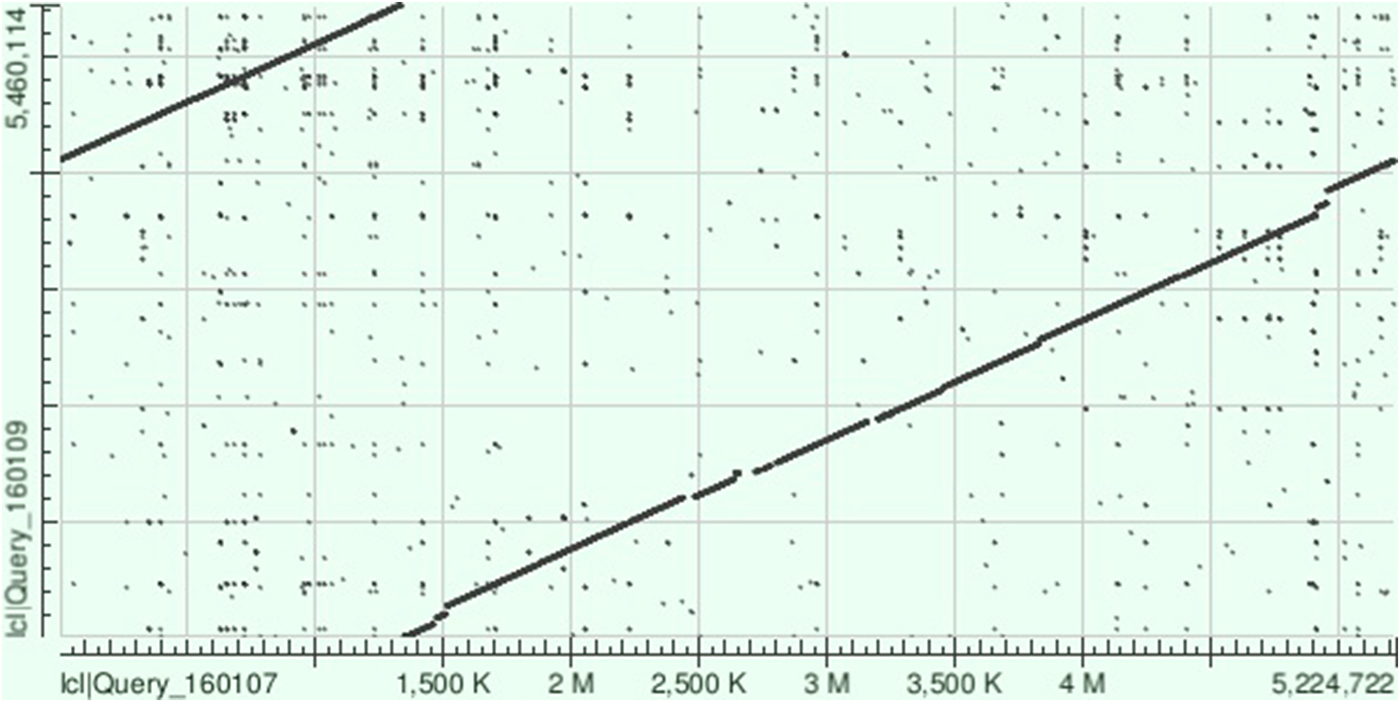
Fig. 2. Dot plot obtained following aligning of the sequences of the chromosomes of KpvK54 (Query_160107) and NCTC 9159 (Query_160109) on BLAST showing the strong similarity between the two sequences and highlighting those relatively small regions of the sequence of the KpvK54 chromosome not present in the NCTC 9159 chromosome sequence (breaks in the line between the two sequences).
However, NCTC 9159 had no virulence plasmid and lacked almost all the virulence genes detected in KpK54, with the exception of the mrkABCDHIJ type 3 fimbrial genes (found in most isolates of K. pneumoniae); there was some evidence for lead resistance genes pbrA and pbrR on the chromosome, but these did not have complete coverage and had low identity (80–89%) with the downloaded version from the MLST website; there was a ferric uptake fur gene. It contained an acquired resistance gene not found in KpvK54 (strAB) but was otherwise similarly devoid of most resistance determinants, with only bla SHV-83, oqxA and oqxB in addition, as in KpvK54. The whole-genome sequence confirmed the capsular type as K39 (wzc_39) and provided the ST of 985, a SLV of ST29 (allelic profile 10,3,2,2,6,4,4 with the gapA allele (underlined) differing from that of ST29). Neither KpvK54 nor NCTC 9159 carried pld1, encoding a phospholipase D family protein, which has also been identified as a virulence factor [Reference Lery25].
Only the chromosome sequence of isolate LS358, of ST29, was available, so we cannot comment on whether it possesses a virulence plasmid or elements such as the aerobactin cluster and tellurite resistance genes associated with them. The only plasmid sequence described of KP-1, also of ST29, (NZ_CP012884) contains none of the virulence elements sought and it therefore likely does not have a virulence plasmid. Similarly to NCTC 9159, the chromosome of the environmental isolate KP-1 lacked most of the virulence genes described among hypervirulent isolates, with the exception of mrkABCDFHIJ; it did not have the yersiniabactin cluster. By contrast, the chromosome of LS358, isolated from liver abscess puncture fluid, carried the yersiniabactin cluster (ybtAEPQSTUX and fyuA, irp1, irp2), but not the virB genes encoding type IV secretion proteins, suggesting that the yersiniabactin cluster was not in an integrative conjugative element (ICE), which requires a type IV secretion system. Surprisingly, we did not detect wcaG in this isolate. Both isolates carried bla SHV-83, oqxA and oqxB resistance genes. As was the case with KpvK54 and NCTC 9159, neither LS358 nor KP-1 carried pld-1.
There were four main regions where the chromosome sequence of KpvK54 had no significant similarity with that of NCTC 9159. Perhaps the most significant of these are the regions of nt 2 656 985–2 718 902, encoding an ‘integrase/retron-type reverse transcriptase, putative ATPase’ (according to the annotations from matches in BLAST) and the virulence-associated yersiniabactin loci (ybtS, ybtX, ybtQ, ybtP, ybtA, irp2 irp1, ybtU, ybtT, ybtE, fyuA), and nt 2 775 659–2 799 701 encoding the capsular polysaccharide (cps) gene synthesis cluster, including wzc_54 and wzi_115. There are three instances of the CCAGTCAGAGGAGCCAA 17 bp flanking direct repeat sequence of K. pneumoniae ICEs (ICEKp) (at nt 2 646 067–2 64 083, 2 656 808–2 656 824 and 271 886–2 718 902) that are associated with mobilisation of yersiniabactin [Reference Lam26] and it is likely that irp1, irp2, fyuA and ybtAEPQSTUX are all within an ICEKp element bounded between nt 2 650 808 and 2 718 902, which covers 96% of the sequence of ICEKp3 and includes an integrase gene and type IV secretory system, essential components of an ICE.
NCTC 9159 was isolated from urine (rather than a site indicating invasive disease) and lacks characteristics associated generally with hypervirulence (including a virulence plasmid), and therefore arguably would not be considered hypervirulent. Similarly, the environmental isolate KP-1 has not been associated with hypervirulence, so the differences between KpvK54 and NCTC 9159 and KP-1 may be significant in this context, particularly where they are also shared with NTUH-K2044 and RJF999, also associated with hypervirulence, and perhaps also with LS358, isolated from liver abscess puncture fluid. The gene matrix showing the number of genes common to each combination of these isolates revealed only 13 coding sequences found in KpvK54, LS358, NUTH-K2044 and RJF999, but not in KP-1 and NCTC 9159, all but two of which belong to the virulence-associated yersiniabactin cluster (ybtS, ybtX, ybtQ, ybtP, ybtA, irp2 irp1, ybtU, ybtT, ybtE, fyuA) (Supplementary Table S3). The remaining genes were an integrase gene (part of the ICEKp element carrying the yersiniabactin cluster) and K54_4379379_4380137 (which partially covers C0077_23090 and RJF9_26025 coding for α-galactosidase/glycoside hydrolase); however, 84% of this coding sequence (corresponding to nt 4 379 379–4 379 969 and nt 4 380 086–4 380 137 of the chromosome of KpvK54) was also present in NCTC 9159 and KP-1. Table 2 gives a list of coding sequences common to KpvK54 and NTUH-K2044 and/or RJF999, but not in NCTC 9159; some of these were also present in LS358 and/or KP-1. NTUH-K2044 and KpvK54 alone of the six genomes shared mobB, mobC, virB11 and ardC. Interestingly, the type IV secretion system genes virB1, virB2, virB3-4, virB5, virB8 and virB10 were common only to KpvK54, NTUH-K2044 and RJF999, and were not found in LS358, nor was alpA2 (encoding a putative transcriptional regulator/DNA-binding protein). There were also a further 26 genes common only to KpvK54 and RJF999, and a further 15 common to KpvK54 and LS358 only (Supplementary Table S3). KpvK54 shared in common with RJF999, a region corresponding to RJF9_19730 to RJF9_19760, which was not present in any of the other genomes. potD, rhaP and eight other genes were shared by all the genomes except NCTC 9159. There were 335 genes common to NTUH-K2044 and RJF999 (K1-ST23 isolates) that were not present in KpvK54, LS358, KP-1 or NCTC 9159 (the CG29 isolates), which included allABCSR (the allantoin cluster), acoABCDK, cas1, cas2, cas3, cas5e, cse1, cse2, cse3, cse4, magA, fdrA, gatABCDYZ, pflCD, sfuABC and ylbCEF. Among the genes found only in K54 isolates, 15 were found in KpvK54 and LS358 that were not present (or in some cases (e.g. C0077_05160) were only partially present) in KP-1 (Supplementary Table S4); there were a further 15 genes found in KpvK54, LS358 and KP-1 that were not present in any of the other genomes (e.g. C0077_09035 and C0077_05470, both coding for hypothetical proteins), only some of which were genes in the capsular polysaccharide synthesis cluster. There were also significant numbers of coding sequences exclusive to each isolate among the six genomes; in the case of the 79 predicted genes found only in KpvK54 these were mainly found together in regions (e.g. 2 709 483–2 717 651 (in the ICE element)) and correspond almost exclusively to ‘hypothetical proteins’ on BLAST searches.
Table 2. Chromosomal genes found in KpvK54 (CP023134) and NUTH-K2044 (AP006725) or RJF999 (CP014010), but not found in NCTC 9159. Those in bold are in an ICEKp element that includes the yersiniabactin cluster. LS358 (CP025629) and KP-1 (CP012883) are representatives of K54-ST29 from liver abscess puncture fluid and the environment, respectively

a nt 2 656 808–2 718 919 in the KpvK54 chromosome corresponds to a ICEKp element closest to ICEKp3.
b Nucleotide positions in KpvK54.
PCR testing showed that RJF9_19740, RJF9_19755 and RJF9_19640 were present exclusively in KpvK54 among the 15 isolates in which they were sought; they were not detected in any of the other three rmpA/rmpA2-positive isolates of K54 (Table 3). C0077_05300, C0077_05255 and C0077_ 04790 were detected in all four rmpA/rmpA2-positive isolates of K54, with C0077_05255 not being found in any of the other isolates tested. These results are consistent with the association of these targets with virulent isolates. Both C0077_05255 and C0077_04790 have few matches on BLAST, indicating that they are not widely found. None of these genes were detected in the four non-CG29 K54 isolates tested.
Table 3. Results of PCR testing for six targets identified as potentially associated with virulence among 11 clonal group 29 (CG29) isolates described in Table 1 (groups A, B and C) and four further isolates of K54 not belonging to CG29

a C0077_0530, C0077_05255 and C0077_04790 were found in the LS358 chromosome, but not in those of KP-1, NCTC 9159, NTUH-K2044 and RJF999.
b RJF9_19740, RJF9_19755 and RJF9_19640 were found in the RJF999 chromosome, but not in those of LS358, KP-1, NCTC 9159 and NTUH-K2044.
c VNTR profiles 4,1,3,2,2,2,2,3,5,2,4 (corresponding to ST530); 6,3,4,2,1,2,3,4,3,3,3 (corresponding to ST240); 1,8,2,4,0,2,2,5,3,2,3 and 1,7,4,15,2,2,1,4,5,2,3.
None of the representatives of the 12 other VNTR types of K54 on our database (from 16 patients) were PCR-positive for rmpA/rmpA2, although they did include seven isolates from blood. Most of the other types were from only one or two patients with the next largest group in the K54 set (after CG29) being from three patients only (in each case from urine) and corresponding to ST530 (VNTR profile 4,1,3,2,2,2,2,3,5,2,4).
Discussion
It is likely that the K54 isolates of CG29 with the virulence plasmid (VNTR profiles 7,3,1,5,1,2,4,1,2/3,2,4) in this study were associated with the hypervirulent phenotype, all being isolated from blood or pus, with clinical details of pyrexia, sepsis or liver abscesses (5/5). Where determined, these isolates all had a repeat number of 2 or 3 at the ninth VNTR locus, in contrast to those without the virulence plasmid, which exclusively had a repeat of 4 at that locus (Table 1). Some of the K54 isolates of CG29 without a virulence plasmid were also isolated from blood and associated with bacteraemia or sepsis (5/10). Similarly, among the non-K54 representatives, there were examples of blood/pus/pleural fluid isolates associated with bacteraemia/abscesses/sepsis, suggesting that the K54 capsular type in itself may not be of great importance, other than that it alerts the investigator to a potentially hypervirulent clonal type. However, it was only isolates of the K54 capsular type that carried the virulence plasmid. While host factors and the clinical procedures carried out will undoubtedly also have been important, these results suggest that the clonal type is a major factor, with the virulence plasmid being an important contributory (but not necessarily essential) factor. It must also be borne in mind that the gastro-intestinal tract is the likely source of the organism in hypervirulent infections, with individuals being colonised prior to infection, so that isolation from screening swabs is not necessarily inconsistent with a hypervirulent type [Reference Paczosa and Mecsas2]. Similarly, it has previously been observed that virulence plasmid-cured Kp52.145 (a hypervirulent strain of capsular type K2) remained more virulent than other strains not carrying the plasmid [Reference Nassif and Sansonetti27], again suggesting that higher pathogenicity is correlated with clonal type, even when the virulence plasmid is absent. However, this goes against observations that suggest that the virulence plasmid is a requirement for hypervirulence, rmpA/rmpA2 and aerobactin, in particular being strongly associated with the hypervirulent syndrome [Reference Hsu10, Reference Tang28, Reference Russo29]. RmpA can be found chromosomally, but it is unlikely that this was the case in this study, since we failed to detect it in our PCRs using two different sets of primers among those isolates with no evidence of a virulence plasmid, nor was it detected in the whole-genome sequences, even searching using the chromosomal-rmpA-specific primers described by Hsu et al. [Reference Hsu10].
However, it would not be true to say that all representatives of CG29, whether of capsular type K54 or not, are associated with virulence, NCTC 9159 (isolated from urine) and KP-1 perhaps being good examples of this, and we therefore set out to compare KpvK54 with NCTC 9159, KP-1, LS358 and hypervirulent representatives of K1-ST23 NTUH-K2044 and RJF999. Despite being remarkably similar to KpvK54, NCTC 9159 lacked the very factors (virulence plasmid, yersiniabactin cluster) that have been identified as important to hypervirulence. Indeed, among the chromosomal genes found in the six isolates we compared, it was almost exclusively the yersiniabactin cluster genes that were common to those most associated with virulence (KpvK54, NTUH-K2044, RJF999 and LS358), but not present in NCTC 9159 and the environmental isolate KP-1. While the yersiniabactin cluster has long been associated with virulence, it is now found commonly in clinical isolates of K. pneumoniae [Reference Turton13, Reference Lam26], so one might expect that other chromosomal elements may also be important. KpvK54 also shared other genes with hypervirulent K1-ST23 isolates (e.g. the mobB and mobC genes encoding mobilisation proteins in the ICE element and a region corresponding to RJF9_19730 to RJF9_19760), not present in NCTC 9159, which may be important. There were also genes found only among the K54 isolates of CG29 among the six genomes compared, some only found among the two K54 isolates associated with virulence (KpvK54 and LS358) (e.g. C0077_05255), which may play a role and provide further explanation for the association of K54 with virulence. Notably, these were all also present in isolate KP10 (CP025091), a further representative of K54-ST29 carrying the yersiniabactin cluster, and were also mostly present in KP14 (CP025093), even when there were few other matches/near-matches. However, we do not know any clinical information concerning these isolates. All three such genes sought by PCR (C0077_05300 (encoding lysozyme), C0077_05255 and C0077_04790) were detected in all four of our rmpA/rmpA2-positive isolates of K54 available for testing. According to InterPro, C0077_05255 is an Ead/Ea22-like protein, belonging to a family containing phage proteins and bacterial proteins likely to represent integrated phage proteins, while C0077_04790 may be involved in bacteriophage resistance. Our approach concentrated on the presence or absence of coding sequences, but changes in those sequences and indeed in non-coding sequences may also have a profound effect on virulence, as evidenced by recent findings with Salmonella enterica serovar Typhimurium ST313 [Reference Hammarlöf30].
Our observations add to the already existing body of evidence identifying CG29 as a hypervirulent type that could explain the association of capsular type K54 with virulence. K54-positive representatives of CG29 were found in similar numbers among our isolates as the hypervirulent ST86 type belonging to K2 (from 14 to 15/4670 patients), although representatives with a virulence plasmid were rarer (from 5/4670 patients). However, the situation is complex; CG29 isolates displayed a range of virulence, from environmental through to hypervirulence, with those most consistently associated with virulence being of capsular type K54 and possessing a virulence plasmid containing both the aerobactin and salmochelin-acquired siderophore systems, showing that it is important to at least characterise isolates for these elements. These all shared a VNTR profile of 7,3,1,5,1,2,4,1,2/3,2,4. Our work suggests that they may also be recognised by the detection of the C0077_05255 marker. ‘Switching’ of capsular type was observed, highlighting that these types may be missed if screened by capsular typing alone.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0950268818001826
Acknowledgements
The authors thank the team of the curators of the Institut Pasteur MLST system (Paris, France) for importing novel alleles, profiles and/or isolates at http://bigsdb.web.pasteur.fr., Oxford Nanopore Technologies for providing a MIN107 flow cell and an SQK-LSK308 1D2 sequencing kit and staff in UK hospitals for sending isolates to the authors. They thank colleagues in AMRHAI for carbapenemase detection and colleagues in the Genomic Services and Development Unit for Sanger sequencing. The authors are also grateful to the NCTC 3000 project team for providing the whole-genome sequence of NCTC 9159.
Financial support
This research received no specific grant from any funding agency, commercial or not-for-profit sectors.







