



Introduction
Epilepsy, a disorder characterized by recurrent, unprovoked seizures, is one of the most common brain conditions worldwide, affecting 7.6 out of 1,000 people in their lifetime. Reference Fiest, Sauro and Wiebe1 Though an array of antiseizure medications (ASM) exists to treat this condition, seizure control is only achieved in this manner in about 70% of individuals, leaving the other 30% with so-called drug-resistant epilepsy. Reference Kalilani, Sun, Pelgrims, Noack-Rink and Villanueva2 Epilepsy represents one of the highest global disease burdens in neurology in both children and young adults. 3 A cause to an individual’s epilepsy can at times be determined; however, an ill-defined number of epilepsies remain of unknown origin and are often presumed to be genetic or autoimmune. Reference Dubey, Alqallaf and Hays4,Reference Thijs, Surges, O'Brien and Sander5
In the last decades, genomic technologies have become increasingly sophisticated, affordable, and accessible. At present, clinicians have access to an assortment of genetic testing options, ranging from genetic panels that can investigate dozens to hundreds of genes, to whole genome sequencing. Reference Katsanis and Katsanis6 Genetic panels are used to diagnose monogenetic disorders by allowing for the analysis of a limited set of predetermined genes. In comparison to whole genome sequencing, panels offer many advantages, notably in terms of costs, running times, and dataset handling. Reference de Koning, Jongbloed, Sikkema-Raddatz and Sinke7 Targeted genetic panels exist for a growing number of medical conditions, including hereditary cancer syndromes, cardiomyopathy, movement disorders, and epilepsy. Reference de Koning, Jongbloed, Sikkema-Raddatz and Sinke7,Reference Tsaousis, Papadopoulou and Apessos8
Whereas genetic testing may once have been considered rather niche, its clinical applications nowadays appear ever widening. Reference Katsanis and Katsanis6 For instance, in epileptology, a better understanding of the genetic underpinnings to many forms of childhood epilepsy has led to important changes in clinical practice. Reference Scheffer9 The indications for genetic testing in pediatric epileptology are various, and the list of genes associated with early-onset epilepsy is rapidly expanding. Reference Hebbar and Mefford10 Many of these epilepsy syndromes have responded to targeted therapies, and thus the idea of “precision medicine” (i.e., tailoring treatments to a patient’s genetic profile) has seemed more and more achievable. Reference Sharma, Hussain and Greenwood11 In the adult setting, however, data regarding the usage of genetic testing in epilepsy is lacking; little is known on the utility, yield, and cost-efficiency of genetic testing in this population. As such, guidelines on genetic testing in the adult epilepsy setting are mostly based on low-quality evidence and expert opinion, and there is a clear need for more data on the subject. Reference Jain, Andrade and Donner12 In this study, we aimed to evaluate the yield and utility of multigene panels in adults with epilepsy in a tertiary care setting.
Methods
Patients
A retrospective, cross-sectional study was performed on consecutive patients followed by an epileptologist at a Canadian academic tertiary care center’s outpatient epilepsy clinic for whom a genetic panel was performed between January 2016 and August 2021. Epilepsy genetic panels were generally ordered for individuals a) who had a diagnosis of epilepsy of unknown etiology or associated with malformations of cortical development and b) who consented to the procedure.
Operational Definitions
A positive family history of epilepsy was determined based only on first-degree relatives. Refractory epilepsy was defined as epilepsy that did not respond satisfactorily to two ASMs, following the criteria put forward by the International League Against Epilepsy. Reference Kwan, Arzimanoglou and Berg13 Epilepsy type was defined as focal-onset versus generalized-onset. Four types of genetic panel findings were possible as determined by the team responsible for the genetic testing: pathogenic variant (PV), likely pathogenic variant (LPV), variant of unknown significance (VUS), and benign variant (BV). One same panel could yield no findings (N), a single finding, or many findings at once. Genetic panel results could be interpreted as positive (i.e., at least one PV or LPV was found), uncertain (i.e., at least one VUS was found with no PV nor LPV), or negative (i.e., the panel was normal, or only BV were found). A positive panel result did not necessarily entail that the patient was diagnosed with a genetic form of epilepsy. A genetic diagnosis could only be made if the patient had two PV or LPV of an autosomal recessive gene, or if the patient had one PV or LPV of autosomal dominant or X-linked inheritance. If a patient had only one autosomal recessive PV or LPV with a phenotype that potentially correlated with a genetic condition, this was considered a positive panel result but not a confirmed genetic diagnosis. In these cases, deletion/duplication analyses were carried out to search for a second pathogenic variant but were negative. The possibility that the patient harbored a second disease-causing pathogenic variant that was undetectable by the deletion/duplication analyses could, however, not be excluded.
Types of Genetic Panels
At our institution, epilepsy-specific genetic panels became more easily accessible for clinical purposes in 2016. Many types of comprehensive epilepsy panels were available throughout the years: Panel A evaluated 144 genes and was available from 2021 onwards; Panel B evaluated 127 genes and was available between 2018 and 2021; Panel C evaluated 87 genes and was available from 2017 to 2018, and panel D evaluated 36 genes and was available from 2016 to 2017. Certain more specialized types of epilepsy panels could be ordered based on clinical suspicion; these included an advanced comprehensive epilepsy panel (panel E) that was available in 2017 and that evaluated 141 genes, a mitochondrial nuclear gene panel (panel F) that evaluated 202 genes, a progressive myoclonic epilepsy panel (panel G) that evaluated 18 genes, and an idiopathic generalized epilepsy panel (panel H) that evaluated 91 genes. The exact genes that were tested in each panel are available in the supplementary material (Supplementary Table S1).
Data Collection and Analysis
Data were collected by manually and retrospectively reviewing panel results and patient notes. Recorded data included the following:
-
1. patient data (identification, age at seizure onset, epilepsy type, whether the epilepsy was refractory, presence of comorbid intellectual disability (ID), presence of a family history of epilepsy, presence of dysmorphisms on physical exam, and presence of a structural brain lesion on imaging),
-
2. and panel data (panel type, the reason for which the panel was ordered, whether the panel was ordered in the context of a preoperative evaluation for epilepsy surgery, panel interpretation, and panel findings, including the gene involved, the coding DNA, the variant, the zygosity, and the clinical significance of the relevant findings).
When a genetic diagnosis was established, whether this diagnosis was clinically actionable (i.e., the diagnosis affected the treatment) was also recorded. Data are presented as means (standard deviation) for continuous variables and count (frequency) for proportions. Descriptive statistical analyses were conducted to synthesize data. Univariate analyses were performed using t tests to determine the association between receiving positive panel results and panel size, as well as between receiving positive panel results and age at seizure onset. All statistical analyses were performed using R version 4.12. 14 Significance level was set at 0.05. Missing data were handled through pairwise deletion.
Results
In total, 164 genetic panels were performed in 164 patients. Table 1 presents the baseline characteristics of this cohort. Mean age at epilepsy onset was 15.5 years ± 11.2 years. Few patients had comorbid ID (10%), a positive family history of epilepsy (11%), and dysmorphisms (1.0%). Most patients had focal epilepsy (86%) and refractory epilepsy (80%).
Table 1: Cohort description
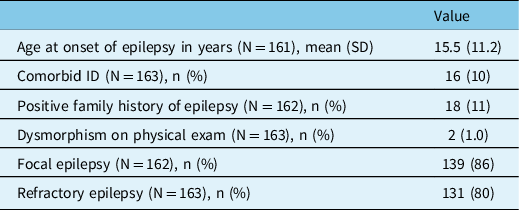
ID = intellectual disability; N = sample size; n = count.
Table 2 presents the types of panels that were ordered. Panel A was ordered 23 times (14%), panel B 79 times (48%), panel C 38 times (23%), panel D 20 times (12%), and panels E through H were each ordered only once (0.6%). Most panels (89%) were ordered for patients with epilepsy of unknown etiology with no structural brain abnormalities; seven panels (4.2%) were ordered for patients with focal cortical dysplasia, six panels (3.7%) for patients with mesial temporal sclerosis, three (1.8%) for patients with multiple sclerosis brain lesions (not thought to be the cause of epilepsy), one panel (0.6%) for a patient with periventricular heterotopia, and one panel (0.6%) for a patient with a temporal meningoencephalocele. Several panels (23%) were performed in the context of a presurgical evaluation.
Table 2: Types of genetic panels ordered

n = count.
Panels A–E were comprehensive epilepsy panels. Panel F was a mitochondrial nuclear gene panel. Panel G was a progressive myoclonic epilepsy panel. Panel H was an idiopathic generalized epilepsy panel.
Figure 1 summarizes the interpretations of the panel results. The yield of positive results was 11% (95% CI 6.6–17%), whereas the proportion of negative results was 40% (95% CI 33–48%). The proportion of uncertain results was 49% (95% CI 41–57%). The yield of genetic diagnoses was 4.3% (95% CI 1.7–8.6%). Of note, out of the 18 positive panels, only seven were considered indicative of genetic diagnoses; the remaining 11 positive panels corresponded to cases in which only one autosomal recessive PV or LPV was found. The seven confirmed genetic conditions were as follows: SCARB2 action myoclonus renal failure syndrome (AMRF), DEPDC5 epilepsy, PCDH19 epilepsy, LGI1 epilepsy, SCN1A epilepsy, MELAS syndrome, and CHRNA7 epilepsy. Of these seven genetic diagnoses, 5 (71%) were evaluated to be clinically actionable: SCARB2 AMRF, DEPDC5 epilepsy, PCDH19 epilepsy, and SCN1A epilepsy. Two diagnoses (DEPDC5 epilepsy and PCDH19 epilepsy) were made in patients who were undergoing a preoperative evaluation for epilepsy surgery. None of the patients with genetic diagnoses had ID. Across the 164 panels, 111 generated 165 findings; otherwise, 53 panels generated no findings. Figure 2 summarizes the panel findings: 12 (7.3%) PV, 6 (3.6%) LPV, 128 (78%) VUS, and 19 (11%) BV.
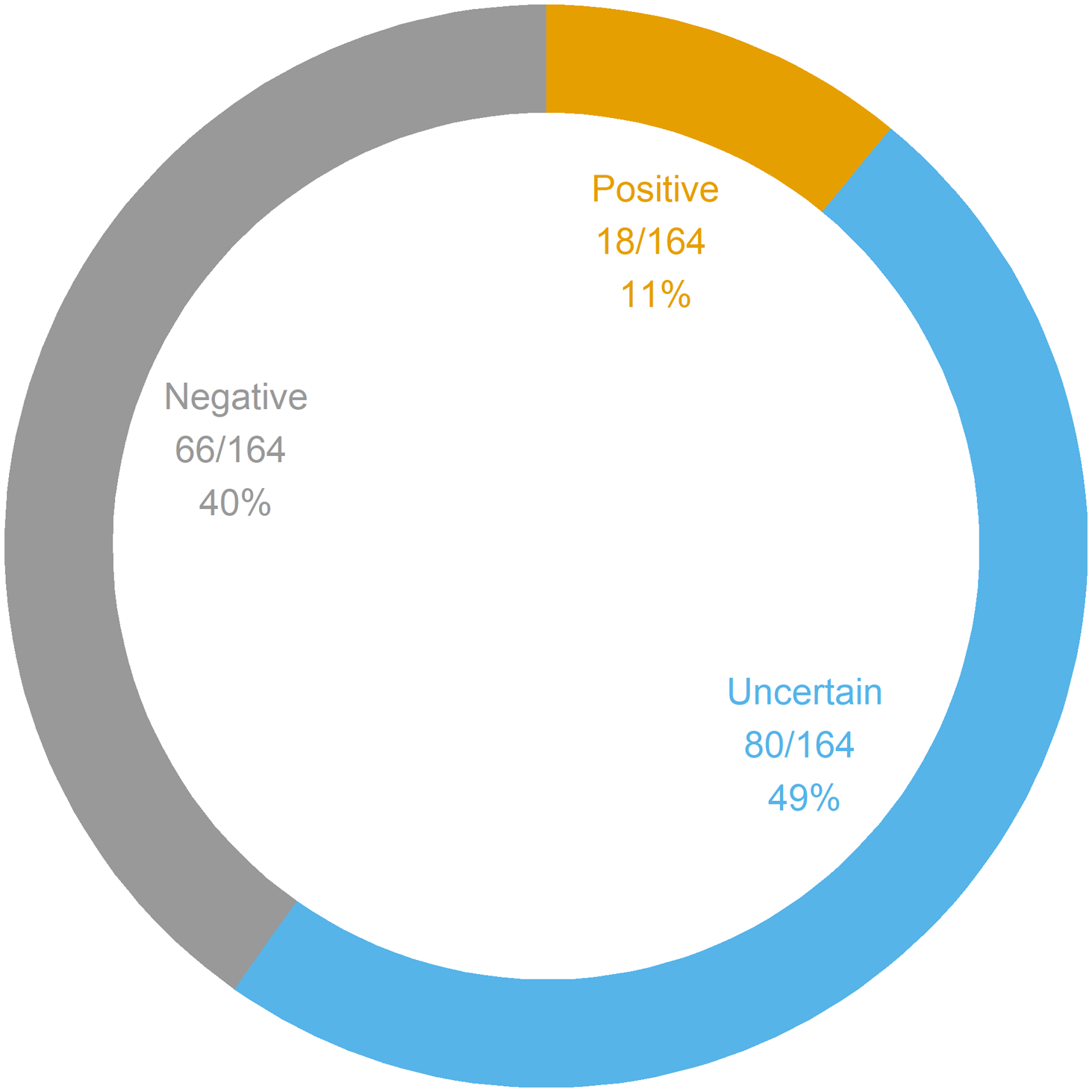
Figure 1: Genetic panel interpretations. A total of 164 panels were ordered in 164 patients. Genetic panel results could be interpreted as positive (i.e., at least one PV or LPV was found), uncertain (i.e., at least one VUS was found with no PV nor LPV), or negative (i.e., the panel was normal, or only BV were found). BV = benign variant; LPV = likely pathogenic variant; PV = pathogenic variant; VUS = variant of unknown significance.
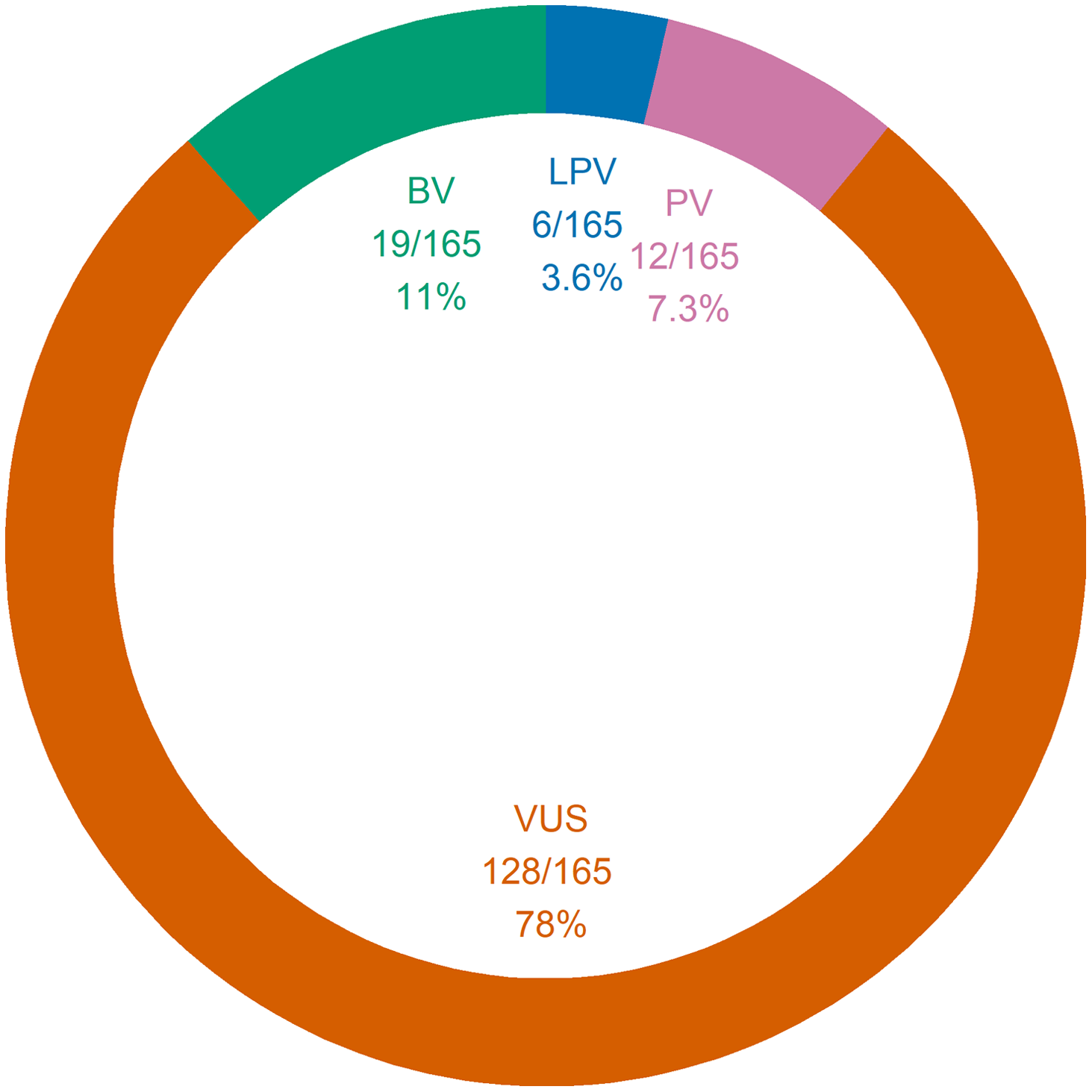
Figure 2: Genetic panel findings. A total of 165 findings were amassed from 111 panels. One same panel could yield a single finding just as it could yield many findings at once. Of note, 53 panels generated no findings. BV = benign variant; LPV = likely pathogenic variant; PV = pathogenic variant; VUS = variant of unknown significance.
Table 3 presents the PV and LPV with the implicated genes, the coding DNA, the zygosity, and the type of panel used. Due to space limitations, a similar but more comprehensive table (Supplementary Table S2) is available in the supplementary material and presents the data regarding PV, LPV, VUS, BV, and negative panels. Also due to space limitations, a summary description of previous studies reporting on genetic panel usage in adults with epilepsy can be found in the supplementary material (Supplementary Table S3).
Table 3: Pathogenic and likely pathogenic variants
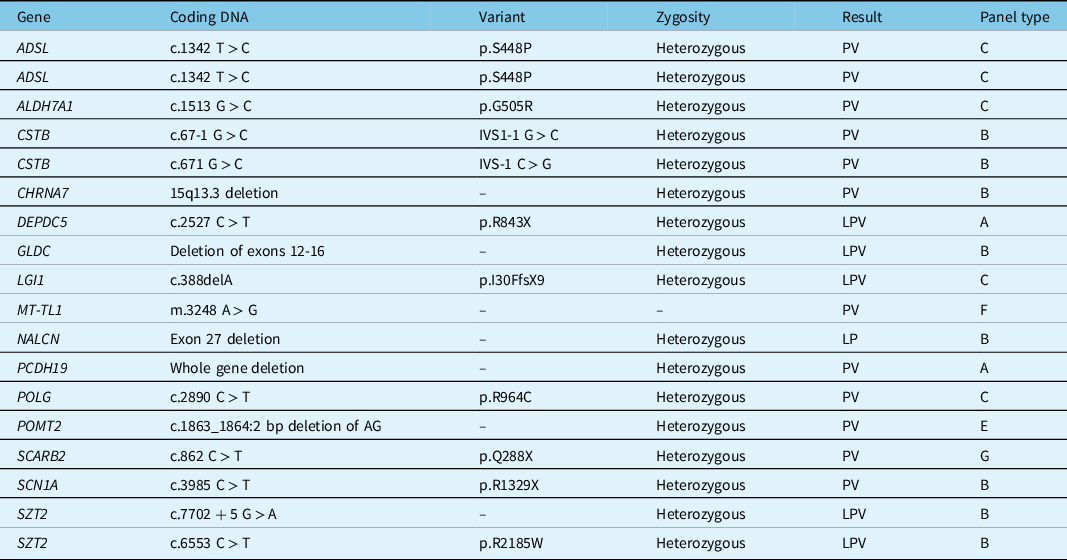
LPV = likely pathogenic variant; PV = pathogenic variant.
The mean number of genes tested by each panel was 106 ± 33 when considering all patients. For patients who received a positive panel interpretation, this mean was 117 ± 36 genes. For patients who received an uncertain or negative panel interpretation, this mean was 105 ± 32 genes. No significant association between panel size and panel interpretation was found (p = 0.193). The mean age at seizure onset was 15.5 ± 11.2 years for all patients, 23.1 ± 13.4 years for patients who received a positive panel interpretation, and 14.6 ± 10.6 years for patients who received an uncertain or negative panel interpretation. Individuals who received a positive panel interpretation had seizure onsets at older ages than individuals who received an uncertain or negative panel interpretation (p = 0.02).
Discussion
In this article, we looked at the usage of genetic panels by an epileptologist in an adult epilepsy clinic over a 6-year period. In the last decades, genetic testing has become more and more affordable and clinically accessible to clinicians. Many options for genetic testing exist and vary in terms of sensitivity, specificity, turnaround time, and cost. Reference Katsanis and Katsanis6 A 2019 meta-analytic cost-effectiveness study of genetic testing strategies in individuals with epilepsy of unknown etiology showed that, after adjusting for potential publication bias, genetic panels were the most cost-effective test, followed by whole exome sequencing. This same publication reported a pooled genetic panel positivity yield of 23%, though most of the patients included in the nine articles used to generate this estimate were children. Reference Sanchez Fernandez, Loddenkemper, Gainza-Lein, Sheidley and Poduri15 A larger 2020 meta-analysis generated a pooled genetic testing positivity yield of 24% in epilepsy, though this yield represented a mix of both genetic panels and exome sequencing, and most included works still reported on pediatric patients. Reference Stefanski, Calle-Lopez, Leu, Perez-Palma, Pestana-Knight and Lal16 In fact, most of the research on genetic testing in epilepsy has so far focused on pediatric samples; the yield of genetic panels in the adult setting has long remained understudied. Studies that do report adult yields often select their patients with strict criteria. For instance, two studies published in the last years reported a genetic panel positivity yield of 22% and of 23% in adults with epilepsy (most of whom also had comorbid ID), respectively. Reference Borlot, de Almeida, Combe, Andrade, Filloux and Myers17,Reference Johannesen, Nikanorova and Marjanovic18 Nevertheless, McKnight et al. very recently published a landmark study evaluating 2,008 adults with epilepsy, with at least 21.8% of whom having comorbid ID, and reported a genetic panel diagnostic yield of 10.9%. Reference McKnight, Bristow and Truty19 To date, this study is the largest to investigate genetic panels results in adults with epilepsy without strict inclusion criteria (such as presence of comorbid ID or of a family history of epilepsy), and its cohort is therefore the most representative of the average adult epilepsy clinic. In comparison, our genetic panel positivity yield was 11%, and our diagnostic yield was 4.3%, a diagnostic yield lower than the 10.9% reported by McKnight et al. This discrepancy can probably be explained through differences in the cohorts and the panels used. Notably, our cohort was smaller and had a lesser proportion of individuals with comorbid ID (10% versus at least 21.8%). In addition, almost none of the genetic panels ordered in this study evaluated more than 150 genes, whereas 56.5% of the panels in McKnight et al.’s study evaluated more than 150 genes. As for the gap between our panel positivity yield and our diagnostic yield, this was due to the fact that patients who had only one autosomal recessive PV or LPV were considered to have a positive panel result but not a confirmed genetic diagnosis.
Obtaining a genetic diagnosis can have a considerable impact on the individual with epilepsy by informing prognosis, limiting additional investigations, aiding in the identification of potential comorbidities, guiding pregnancy counseling, bringing a sense of closure to the families involved, and even informing treatment options. Reference Jain, Andrade and Donner12,Reference Ottman, Hirose and Jain20 In our study, though only seven genetic diagnoses were emitted, five were clinically actionable, meaning that they had tangible consequences on the patient’s treatment. In SCARB2 AMRF, affected individuals should benefit from close renal function surveillance, and carbamazepine, oxcarbazepine, and lamotrigine should be avoided. Reference Amrom, Andermann, Andermann, Adam, Ardinger, Pagon and tal21 In DEPDC5 epilepsy, focal cortical dysplasia should be searched for potential epilepsy surgery while a potential role for mTORC1 inhibitors is under investigation. Reference Baulac, Weckhuysen, Adam, Ardinger and Pagon22,Reference Myers and Scheffer23 Clobazam and bromide have been suggested to be the most effective medications in the often refractory PCDH19 epilepsy. Reference Samanta24 In SCN1A epilepsy, sodium channel blockers should be avoided, epilepsy surgery should probably not be pursued unless the phenotype is mild, and a potential use of cannabidiol, stiripentol, fenfluramine, and clemizole in the treatment of this condition is under investigation. Reference Musto, Gardella and Moller25–Reference Strzelczyk and Schubert-Bast28 In MELAS syndrome, multidisciplinary care is crucial, and valproic acid as well as many other ASMs should be avoided due to concerns of mitochondrial toxicity. Reference El-Hattab, Adesina, Jones and Scaglia29 Whereas in McKnight et al.’s study 55.5% of genetic diagnoses were clinically actionable, in our study, 71% of diagnoses were clinically actionable. This discrepancy is probably due to differences in sample sizes, as McKnight et al.’s study reported 218 genetic diagnoses, whereas we only reported seven. Reference McKnight, Bristow and Truty19
On a separate note, despite that many genetic diagnoses may indeed lead to changes in management, the real utility of genetic panels in the adult epilepsy setting is unsure, as confirmed diagnoses remain uncommon. Most panels, both in McKnight et al.’s study and in our study, yielded uncertain or negative results and consequently were not considered to have had any tangible, treatment-modifying impact. Still, the actual utility of genetic panels in the adult epilepsy setting is more nuanced, as even negative panels may theoretically have some impact on individuals by, for instance, conveying a sense of closure. Uncertain panel results, which were the most frequent results, could also hypothetically affect management on a case-by-case basis, though according to the ACMG guidelines, a VUS should not be used in clinical decision-making. Reference Richards, Aziz and Bale30 Nevertheless, because they are predominantly found in sequencing-based genetic tests (49% in our study), VUS pose a significant clinical challenge. VUS that are identified from a targeted genetic panel (rather than from exploratory WES) can often be a good fit with a patient’s clinical presentation; in these cases, further in vitro and/or in vivo functional testing could help confirm, exclude, or at least guide clinicians toward a diagnosis. Functional characterization is usually performed in research laboratories using different in vitro models like biochemical assays and patient-derived cells or using in vivo animal models. Although the latter can be considered as the gold standard of functional characterization, in vivo models remain expensive and time-consuming. The development of a platform for the functional characterization of VUS has been shown to be very successful in the field of oncology (particularly for breast cancers associated with mutations in BRCA genes), leading to a significant improvement in the clinical management of cancer patients. Reference Woods, Baskin and Golubeva31–Reference Federici and Soddu33 Interestingly, a recent work using nematodes described an in vivo platform for screening the pathogenicity of VUS in the STXBP1 gene, which is associated with epileptic syndromes. Reference McCormick, Brock and Wood34 This illustrates how simple in vivo assays could quickly and accurately determine if a VUS is pathogenic or benign. Based on our data, it is worth noting that if only 10% of the VUS identified in our cohort could be functionally validated (a rather conservative estimate), this would have the potential of increasing by 70% the overall positivity yield of our genetic testing. Thus, the clinical advantages of genetic testing can be multiplied via the concomitant development of approaches that aim at functionally validating VUS. Further analysis of the utility of VUS was however considered beyond the scope of this study.
Currently, there are no up-to-date international guidelines on genetic testing in adults with epilepsy. A report on genetic testing in epilepsy from the ILAE Genetics Commission was last published in 2010. Due to how rapidly genetic research is taking place, instead of offering specific recommendations, the ILAE Genetics Commission offered a general framework for evaluating the utility of genetic testing in various situations. It was nonetheless established at that time that although many genes had been identified for epilepsy syndromes, few were very clinically useful to test for. Much has changed since 2010, notably in terms of the rising accessibility of genetic testing and the discovery of a plethora of new genes associated with epilepsy. Reference Katsanis and Katsanis6,Reference Wang, Lin and Liu35 A set of guidelines on genetic testing in epilepsy was released in 2019 by a Genetic Testing Advisory Committee in Ontario, Canada. This publication represents to our knowledge the most recent guidelines on the topic in North America. Suggested indications for genetic testing in epilepsy were as follows: a) clinically consistent with a distinct epilepsy syndrome, b) poor prognosis or high likelihood of lethal outcome, c) refractory epilepsy with no apparent acquired cause, d) suggestive of inborn errors of metabolism, e) associated with malformations of cortical development, f) associated with paroxysmal neurological features (e.g., hemiplegic migraine), g) associated with syndromic features such as ID or dysmorphic features, and h) positive family history of epilepsy. Reference Jain, Andrade and Donner12 According to these guidelines, a comprehensive gene panel can be ordered when a distinct epilepsy syndrome/group is not suspected (otherwise, a focused gene panel may be preferable). Furthermore, genetic testing should not proceed without the ability to ensure pre- and post-test genetic counseling. Reference Jain, Andrade and Donner12 In our practice, we were probably more liberal in ordering panels than what was recommended by the Ontarian guidelines, since we proposed a panel to all adults with epilepsy of unknown etiology. Nevertheless, most of these individuals had refractory epilepsy, and several individuals had focal cortical dysplasia, comorbid ID, or a positive family history of epilepsy. About a quarter of our panels were ordered in the context of a presurgical evaluation, a scenario that was not discussed in the Ontarian guidelines. Surprisingly little is known about the value of genetic testing in the preoperative workup for epilepsy surgery. However, a 2018 systematic review of surgical outcomes in patients with genetic epilepsies showed that surgery was almost never effective in epilepsies caused by channelopathies and disorders of synaptic transmission (with perhaps the exception of SCN1B epilepsy) but was often effective in epilepsies caused by mTOR pathway mutations. Reference Stevelink, Sanders and Tuinman36 The preoperative evaluation for epilepsy surgery is complex, and though genetic testing may eventually be shown to be a useful modality for determining surgical candidacy, it remains unknown how genetic findings should be interpreted in light of other preoperative findings. In our cohort, two genetic diagnoses were confirmed in individuals who are, as of this study’s redaction, still undergoing their presurgical evaluation. For the individual with DEPDC5 epilepsy, this genetic diagnosis could be used as an argument in favor of surgery, as epilepsies caused by mTOR pathway mutations seem to respond well to surgical interventions. Reference Stevelink, Sanders and Tuinman36 For the individual with PCDH19 epilepsy, though a few successful surgeries have been reported for this condition, the evidence for or against surgery remains scarce. Reference Samanta24 Otherwise, none of the patients featured in our cohort were determined to be inadequate surgical candidates on basis of genetic results alone, though our panel yield and sample size were clear limiting factors in this analysis. As for the ability to ensure timely pre- and post-test genetic counseling to patients, integrating a genetic counselor to the epilepsy clinic would probably be the optimal solution. More data is necessary to investigate the utility of genetic testing in the preoperative workup for epilepsy surgery and the practical impact of integrating a genetic counselor in the epilepsy clinic.
Proposing recommendations for genetic testing in epilepsy is difficult considering the speed at which the realm of genetics is advancing. In the upcoming years, it is predicted that the cost of genetic testing will continue to fall; eventually, whole exome sequencing and whole genome sequencing may become more cost-effective than genetic panels. More and more companies are commercializing genetic testing, many of which are already building business models around offering whole exome sequencing and whole genome sequencing directly to the patient. Reference Wolff and Wolff37 New epilepsy genes are continually being discovered, and there is no evidence that this trend will soon end. The landscape of genetic testing is consequently in constant flux, and whatever recommendations exist today may be modified in the future. Nevertheless, perhaps precisely because genetic testing is advancing at such a rapid pace, there is a dire need for regularly updated recommendations to guide clinicians, including those in adult epilepsy clinics. To do so, more data need to be gathered on genetic testing in adults with epilepsy, and we hope our study may encourage future research on this understudied topic.
This study featured several limitations. Firstly, we had little control over which epilepsy panel was ordered; the choice was made by our institution’s genetic medicine service based on provincial reimbursement criteria, cost efficiency, the suspected genetic condition, estimated processing delays, etc. Providers also changed over time, and, as such, our study featured different types of genetic panels that evaluated varying sets of genes (although many genes were common amongst panels – see Supplementary Table S1). Secondly, panels were only ordered in individuals who consented. Thirdly, an unknown but probably growing number of patients who we inherited from pediatric neurologists over the years had already undergone genetic panels. Since a repeat panel would not have been necessary for those with a positive genetic finding, this could have had a negative impact on the diagnostic yield we report here. This may also explain why positive panel interpretations were associated to an older age at seizure onset, since patients who were younger at seizure onset and who had previously undergone a genetic panel with positive results were probably less represented in our cohort. Fourthly, tissue analyses to evaluate for somatic mutations were not carried out in this study, which could once more decrease our yields. Finally, none of the VUS were revaluated to see if they could be reclassified into PV or BV.
Conclusion
In this retrospective, cross-sectional study of genetic panels in the adult epilepsy clinic, a yield of positive panel results of 11% was obtained. Genetic diagnoses were confirmed in 4.3% of patients, with most of these diagnoses affecting in some way the patient’s management. Currently, guidelines on genetic testing in epilepsy are mostly based on low-quality evidence and expert opinion, and there is a clear need for more data on the subject, especially in the adult setting. Future avenues of research include investigating the utility of genetic testing in the preoperative workup for epilepsy surgery and the impact of integrating a genetic counselor to the adult epilepsy clinic.
Acknowledgments
DKN holds a Canada Research Chair in Epilepsy.
Declaration of Interests
The authors have no conflict of interest to disclose.
Statement of Authorship
JL: study conception, data collection, data analysis, drafting, corresponding author; DHT: study conception, manuscript revision, supervision; ML: manuscript revision; MT: manuscript revision; PC: manuscript revision; ES: manuscript revision; DKN: study conception, data analysis, manuscript revision, supervision, principal investigator.
Supplementary Material
To view supplementary material for this article, please visit http://doi.org/10.1017/cjn.2022.49.








 Open access
Open access