



Introduction
Bjarebyite-group minerals are secondary phosphates that occur primarily in granitic pegmatites, but have also been found in the oxidised zones of ore deposits. Bjarebyite was first described from the Palermo No.1 pegmatite, New Hampshire, USA by Moore et al. (Reference Moore, Lund and Keester1973) and its atomic structure was determined by Moore and Araki (Reference Moore and Araki1974). The bjarebyite group currently consists of seven International Mineralogical Association (IMA) approved minerals (Table 1). The minerals have the general formula XM12M22(PO4)3(OH)3 where X = Ba, Sr, Pb; M1 = Fe2+, Mn2+, Mg and M2 = Al or Fe3+. Until the discovery of strontioperloffite (Elliott, Reference Elliott2019), all group members contained Ba as the dominant cation at the X site. The new mineral species, plumboperloffite, characterised and described in this paper, is the first member of the group with Pb dominant at the X site. The name is for the relationship to perloffite with Pb dominating the X site. The mineral and mineral name (symbol Pplf) have been approved by the International Mineralogical Association, Commission on New Minerals, Nomenclature and Classification (IMA2020-007, Elliott and Kampf, Reference Elliott and Kampf2020). The type specimen is deposited in the South Australian Museum, Adelaide, South Australia, (Registration number G34868).
Table 1. Comparison of related minerals.

Occurrence
Plumboperloffite was found on a specimen collected in 2018 from the dumps of the Wiperaminga Hill West Quarry, Boolcoomatta Reserve, Olary Province, South Australia, Australia (140°27'34''E, 31°57'42''S). Pegmatites at Wiperaminga Hill were exploited for feldspar, beryl and muscovite between 1957 and 1980 (Olliver and Steveson, Reference Olliver and Steveson1982). Four pegmatites were worked in three workings on Wiperaminga Hill West and Wiperaminga Hill East. Recorded production is 31.7 tonnes of beryl and 2030 tonnes of feldspar. The pegmatites occur near the base of a prominent range of hills which comprise banded quartz–mica gneiss and schist of the Early to Middle Proterozoic age Willyama Complex. The metasediments have been granitised and in many places are intruded by granite and associated pegmatite and aplite bodies. The pegmatites are usually small lenticular bodies varying from 6 m to 30 m in length and from 1 m to 3 m in width (Hiern, Reference Hiern1966). The pegmatites are mineralogically zoned and comprise an outer border zone of fine- to medium-grained microcline, quartz, plagioclase and muscovite, an intermediate zone of coarse-grained muscovite, quartz, microcline, plagioclase, beryl and apatite (and triplite), and an inner quartz core or cores (Lottermoser and Lu, Reference Lottermoser and Lu1997). The Wiperaminga Hill pegmatites belong to the beryl–columbite–phosphate rare-element type in the classification of Černý (Reference Černý1991). Phosphate minerals, dufrénite and florencite, were first reported from Wiperaminga Hill by King (Reference King1954), and Hiern (Reference Hiern1966) reported apatite, triplite, dickinsonite and barbosalite. During a visit to the locality in 2018 by one of the authors (PE), masses of orange to dark brown triplite–zwieselite were found on the dumps of the eastern quarry. Secondary phosphate minerals have formed in seams and cavities in the triplite–zwieselite by hydrothermal alteration and weathering, in an oxidising, low-temperature, low-pH environment (Lottermoser and Lu, Reference Lottermoser and Lu1997). Plumboperloffite occurs in a single cavity in a matrix comprising triplite, barbosalite and minor quartz. Associated minerals are fluorapatite, phosphosiderite, natrodufrénite and fluorite.
Physical and optical properties
Plumboperloffite occurs as thin tabular to bladed crystals, with a brownish-orange colour. Individual crystals are up to 40 μm in length (Fig. 1). The streak is pale orange and the mineral has a vitreous lustre. The hardness is estimated to be 4 based on the hardness of other bjarebyite-group minerals. The fracture is uneven, the tenacity is brittle and the mineral has an excellent cleavage on {100}. Density was not determined owing to the small amount of material available. The calculated density is 4.416 g/cm3 from the empirical formula. Plumboperloffite is optically biaxial (+) with indices of refraction α = 1.87(1), β = 1.88(1) and γ = 1.89(1) measured in white light. The 2V measured directly by conoscopic observation is 88(1)°. The calculated 2V is 90.5°. Dispersion is apparently strong, based on extinction colours, but the sense could not be determined because of the indistinct conoscopic figure. The pleochroism in shades of yellow brown is X < Z < Y. The optical orientation is Y = b. The optical orientation could not be completely determined because crystals are subparallel intergrowths and the refractive indices are high. The compatibility index 1 – (K P/K C) is 0.03 (excellent) based upon the empirical formula, density calculated using the single-crystal cell and the measured indices of refraction (Mandarino, Reference Mandarino1981).

Figure 1. Orange crystals of plumboperloffite associated with natrodufrénite (black) and fluorite (white). The scale bar is 200 mm. South Australian Museum registration number G34868.
Chemical composition
Analyses of plumboperloffite (12 spots) were performed with a Cameca SXFive electron microprobe (wavelength-dispersive spectroscopy mode, 20 kV, 20 nA and 5 μm beam diameter). Data were processed using the φ(ρZ) correction procedure of Pouchou and Pichoir (Reference Pouchou, Pichoir, Heinrich and Newbury1991). There was insufficient material for direct determination of H2O content so it was based on the crystal structure analysis. The results of the analyses are reported in Table 2. The empirical formula unit, based on 15 O atoms per formula unit (apfu) is (Pb0.92Ca0.04Ba0.01K0.01)Σ0.98(Mn2+1.84Fe2+0.13)Σ1.97(Fe3+1.97Al0.03)Σ2.00(P3.01O11.94)(OH)Σ3.06. The ideal formula is PbMn2+2Fe3+2(PO4)3(OH)3 which requires PbO 29.19, MnO 18.55, Fe2O3 20.87, P2O5 27.86, H2O 3.53, total 100.00 wt.%.
Table 2. Analytical data for plumboperloffite.

* FeO and Fe2O3 calculated to give Fe3+ + Al = 2.00
** H2O calculated from the refined formula.
Infrared spectroscopy
The infrared spectrum of plumboperloffite (Fig. 2) was recorded using a Nicolet 5700 FTIR spectrometer equipped with a Nicolet Continuum IR microscope and a diamond-anvil cell. Data were acquired from a powdered sample during 50 scans in the wavenumber range 4000 to 700 cm–1. The spectrum shows bands at 3471 cm–1 and 3209 cm–1 that correspond to O–H-stretching vibrations of OH groups forming weak hydrogen bonds (Libowitzky and Beran, Reference Libowitzky, Beran, Beran and Libowitzky2004). The bands at 1157, 1067, 989 and 898 cm–1 are assigned to ν3 vibrations of the PO4 tetrahedra and the band at 827 cm–1 is assigned to the ν1 vibration of the PO4 tetrahedra. No absorption attributed to H–O–H bending vibrations was observed in the spectrum.

Figure 2. The Fourier-transform infrared spectrum of powdered plumboperloffite.
Crystallography
Powder X-ray diffraction
Powder X-ray diffraction data for plumboperloffite were obtained using a Rigaku R-AXIS Rapid II curved-imaging-plate microdiffractometer, with monochromatised MoKα radiation (50 kV and 40 mA). A Gandolfi-like motion on the φ and ω axes was used to randomise the sample. Observed d values and intensities were derived by profile fitting using JADE 2010 software (Materials Data, Inc.). Data (in Å for MoKα) are given in Table 3. Unit-cell parameters refined from the powder data using JADE 2010 with whole-pattern fitting are a = 9.17(2), b = 12.41(2), c = 5.02(2) Å, β = 100.84(5)° and V = 561(3) Å3.
Table 3. Powder X-ray data (d in Å) for plumboperloffite.*
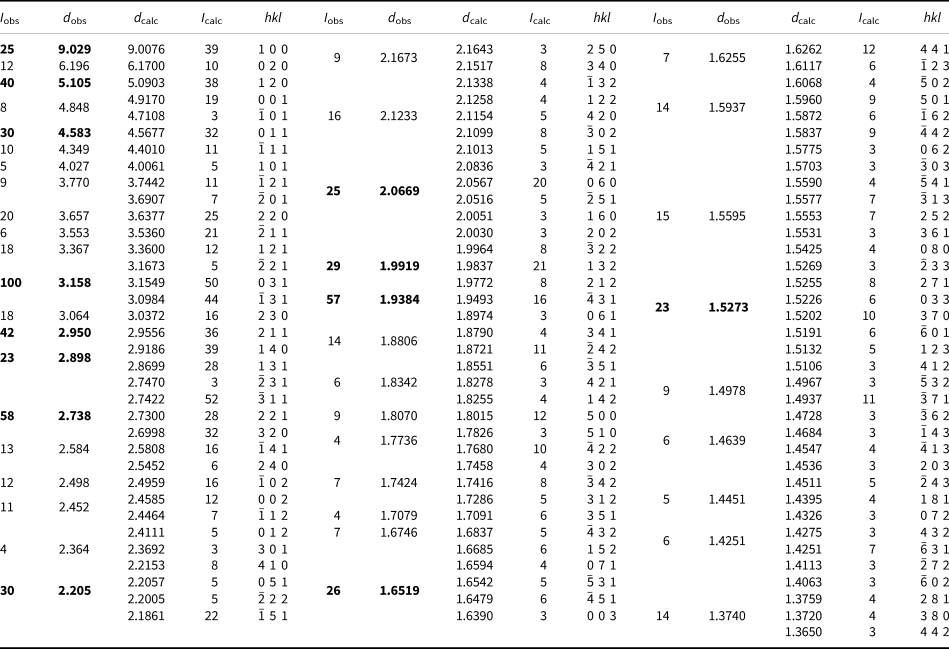
* Only calculated lines with I ≥ 3 are listed. I calc values have been scaled so that the three lines contributing to the highest observed peak have a total intensity of 100; the strongest lines are in bold.
Single-crystal X-ray diffraction
The single-crystal X-ray diffraction experiment was carried out on the micro-focus macromolecular MX2 beamline at the Australian Synchrotron, part of ANSTO (Aragao et al., Reference Aragao, Aishima, Cherukuvada, Clarken, Clift, Cowieson, Ericsson, Gee, Macedo, Mudie, Panjikar, Price, Riboldi-Tunnicliffe, Rostan, Williamson and Caradoc-Davies2018). A crystal was attached to a MiTeGen polymer loop and X-ray diffraction data were collected at 100 K using a Dectris EigerX 16M detector and monochromatic radiation with a wavelength of 0.710754 Å. The intensity data sets were processed using XDS (Kabsch, Reference Kabsch2010) without scaling and with absorption correction and scaling performed using SADABS (Bruker, Reference Bruker2001). Crystal data and experimental details are reported in Table 4.
Table 4. Crystal data, data collection and refinement details.

† wR 2 = Σw(|Fo|2–|Fc|2)2 / Σw|Fo|2)1/2; w = 1/[σ2(Fo2) + (0.0646P)2 + 2.08P]; P = ([max of (0 or F 02)] + 2Fc2)/ 3
Structure refinement
The structure was solved in space group P21/m using SHELXT (Sheldrick, Reference Sheldrick2015a) as implemented in the WinGX suite (Farrugia, Reference Farrugia2012). SHELXL-2018 (Sheldrick, Reference Sheldrick2015b) was used for the refinement of the structure. Following the solution of the structure, the atom coordinates were transformed to correspond to those in the structure of perloffite (Elliott and Willis, Reference Elliott and Willis2011). Difference-Fourier syntheses then located all H atom positions. The H sites were refined with soft restraints on the O–H distances of 0.85(3) Å for the O–H groups and 0.90(3) Å. The final cycle of the refinement converged to R 1 = 0.0207. Final atom coordinates and anisotropic-displacement parameters are listed in Table 5, selected interatomic distances and framework angles are given in Table 6, and bond-valence values, calculated using the parameters of Gagné and Hawthorne (Reference Gagné and Hawthorne2015) are given in Table 7. The crystallographic information file has been deposited with the Principal Editor of Mineralogical Magazine and is available as Supplementary material (see below).
Table 5. Fractional atomic coordinates and displacement parameters for plumboperloffite.

Table 6. Selected interatomic distances (Å), angles (°) and hydrogen bonds for plumboperloffite.

Table 7. Bond-valence* analysis for plumboperloffite.

* Bond-valence parameters used are from Gagné and Hawthorne (Reference Gagné and Hawthorne2015). Bond-valences for the X, M1 and M2 sites are based on the refined occupancy.
Structure description
Plumboperloffite is isostructural with minerals of the bjarebyite group. The structure contains one X site (occupied by Pb and minor Ca, Ba and K), one M1 site (occupied by Mn2+ and minor Fe2+), one M2 site, (occupied by Fe3+ and minor Al) and two P sites. The M1 and M2 sites were refined with full occupancy of Mn and Fe, respectively.
[M2O3(OH)3] octahedra share edges to form [M 2O6(OH)4] dimers which corner-link to form (M22Φ9) chains along the b-direction (Fig. 3). Octahedra that share corners are also linked by P1O4 tetrahedra. The chains are decorated by P2O4 tetrahedra, which link between chains in the c-direction via the O5 and O6 anions. Chains are linked in the a-direction via [M1O5(OH)] octahedra, which share an edge with [M23+O3(OH)3] octahedra and corners with P1O4 and P2O4 tetrahedra (Fig. 4). The X site occupies channels parallel to [001] and also provides linkage between chains in the a-direction. The X site exhibits a lop-sided coordination (Fig. 5) with three very short bonds to O1 and O7, four medium length bonds to O2, O4 and OH8, and six very long bonds to O5, O6 and O7. This coordination is distinct from the more regular coordination of the X site in the structures of other bjarebyite-group minerals and is attributable to the accommodation of the stereoactive 6s2 electron lone pair on Pb2+. Coordination of the M1 and M2 sites is similar to the sites in the structures of other bjarebyite-group minerals. The M1O6 octahedron is strongly distorted with bond angles in the range 86.35 to 109.53°. The M1O6 octahedron is relatively undistorted with bond angles in the range 86.77 to 96.22°.

Figure 3. The crystal structure of plumboperloffite projected on (100). Hydrogen atoms are grey spheres. Hydrogen bonds are indicated by dotted lines. Structure drawings were completed using ATOMS (Shape Software, Kingsport, Tennessee, USA).

Figure 4. The crystal structure of plumboperloffite projected onto (001). The X positions are shown as purple spheres. The unit cell is outlined.

Figure 5. The Pb coordination in plumboperloffite showing Pb–O bond lengths (in Å).
The hydrogen bonding scheme for plumboperloffite given in Table 6, is the same as reported in previous studies on bjarebyite-group minerals (e.g. Cooper and Hawthorne, Reference Cooper and Hawthorne1994; Elliott, Reference Elliott2019). The H9 atom associated with OH9 hydrogen bonds to O3 with a donor–acceptor distance of H9⋅⋅⋅O3 = 1.89(3) Å and an OH9–H2⋅⋅⋅O3 angle of 168(8)°, an arrangement typical of strong hydrogen bonding (Libowitzky and Beran, Reference Libowitzky, Beran, Beran and Libowitzky2004). The H8 atom provides a weak hydrogen bond to O6 with a donor–acceptor distance of H8⋅⋅⋅O6 = 2.73(2) Å and an OH8–H8⋅⋅⋅O6 angle of 124(6)°.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1180/mgm.2023.97.
Competing interests
The authors declare none.
Acknowledgements
The authors thank Ben Wade of Adelaide Microscopy, The University of Adelaide for assistance with the microprobe analysis. The infrared spectrum was acquired with the assistance of the Forensic Science Centre, Adelaide. This research was undertaken in part using the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector.















 Open access
Open access