


Introduction
In the last decade, CVD has become the leading cause of mortality and morbidity, with increasing prevalence worldwide(Reference Perk, De Backer and Gohlke1,Reference Shay, Gooding and Murillo2) . One of the main causes in the pathogenesis of CVD is impairment of cholesterol homeostasis(Reference Kisioglu and Nergiz-Unal3). Cholesterol and lipoprotein homeostasis is influenced by the endogenous synthesis of cholesterol, absorption of dietary cholesterol in the gastrointestinal tract, transport of cholesterol via lipoproteins in the circulation, and the reabsorption of cholesterol excreted in the form of bile acids. Likewise, recent studies have noted that the levels and/or expressions of enzymes, transporters, receptors and transcription factors involving in cholesterol metabolism are influenced by dietary fat and fatty acids(Reference Kisioglu and Nergiz-Unal3-Reference Ooi, Watts and Ng6).
The amount of dietary total fat, fatty acids and fatty acid pattern are important for cholesterol homeostasis and CVD. Dietary fatty acids affect cholesterol homeostasis in different ways depending on the existence of double bonds and their cis or trans forms(Reference Eckel, Jakicic and Ard7). Under these circumstances, high dietary SFA and trans-fatty acids (TFA) might cause dyslipidaemia and hypercholesterolaemia by impairing cholesterol metabolism(Reference de Oliveira Otto, Mozaffarian and Kromhout8,Reference Gebauer, Destaillats and Dionisi9) . On the contrary, it has been reported that a diet high in MUFA might be protective against CVD by improving cholesterol and lipoprotein homeostasis(Reference Arapostathi, Tzanetakou and Kokkinos10). Though the effects of total PUFA, n-3 PUFA and n-6 PUFA content in the diet on cholesterol homeostasis are still controversial, the amount of total PUFA, n-6 PUFA and n-3 PUFA, and the n-3:n-6 PUFA ratio are important for cholesterol homeostasis(5,Reference Ooi, Watts and Ng6) .
Although some of the mechanisms by which dietary fatty acids affect cholesterol metabolism are clearly known, novel mechanisms that may influence cholesterol metabolism are also argued in the literature(Reference Ooi, Watts and Ng6,Reference Lamarche and Couture11) . One of the proposed novel mechanisms is the function of CD36 (fatty acid translocase/cluster of differentiation 36) as a fatty acid transporter, but there are a limited number of studies clarifying this role in cholesterol homeostasis(Reference Nigro, Menotti and Cento12,Reference Wu, Sarna and Hwang13) . Dietary fatty acids might influence CD36-mediated cholesterol metabolism by altering CD36 levels and/or expression in macrophages, liver, small intestine, and some other cells and tissues involved in cholesterol homeostasis(Reference Nigro, Menotti and Cento12,Reference Buttet, Poirier and Traynard14,Reference Cedo, Metso and Santos15) . All this information highlights that the CD36 receptor is thought to be a novel nutrient-sensitive biomarker for cholesterol homeostasis.
Nowadays, there is an increasing number of studies related to the effect of dietary fatty acids on cholesterol metabolism induced by CD36 in the literature(Reference Rodrigue-Way, Caron and Bilodeau16,Reference Maréchal, Laviolette and Rodrigue-Way17) . However, there is a lack of knowledge in combining the potential mechanisms of dietary fatty acids on CD36-mediated cholesterol metabolism. Thus, the novel nutritional target of CD36 and interventions that focus on dietary fatty acids on potential mechanisms underlying cholesterol homeostasis need to be discussed. Therefore, the present review is written to reveal the novel effects of dietary fatty acids on CD36-mediated cholesterol homeostasis in terms of absorption, endogenous synthesis, circulation and excretion of cholesterol.
Method of literature search
The literature search was performed by using the databases of PubMed, Science Direct, Google Scholar and Scopus with the keywords ‘dietary fatty acid types and CD36 and/or the CD36 related mechanisms of cholesterol metabolism’. Reviews, systematic reviews, meta-analyses, epidemiological studies, randomised controlled trials and experimental studies conducted on human subjects, animals and cell cultures were included from the relevant literature. Articles in the English language and published between 2000 and 2019 were involved in the present review.
Structure and functions of CD36
CD36, of which many functions have been discovered in recent years, was first found in platelets and was named platelet glycoprotein IV (GPIV). After that, CD36 was defined as fatty acid transporter (FAT) and receptor in macrophages for oxidised LDL (oxLDL)(Reference Febbraio and Silverstein18,Reference Nergiz-Unal, Rademakers and Cosemans19) . In spite of its simple molecular structure, the transmembrane domain provides receptor properties in addition to its carrier protein property. Additionally, CD36 is a part of the class B scavenger receptor family along with scavenger receptor class B tip I (SR-BI) and lysosomal integral membrane protein II (LIMP II)(Reference Febbraio and Silverstein18,Reference Nergiz-Unal, Rademakers and Cosemans19) . The molecular weight of CD36 ranges from 80 to 90 kDa and has two transmembrane domains (Fig. 1). First, it was determined that the amino acid sequence 93–120 is the binding site for thrombospondin (TSP) 1 and TSP2. Subsequently, it was revealed that the binding site located in amino acid sequence 155–183 is for oxLDL, advanced glycation endproducts and apoptotic cells, sequence 146–164 is for Plasmodium falciparum-infected erythrocytes, and sequence 127–279 is for long-chain fatty acids (LCFA)(Reference Febbraio and Silverstein18-Reference Martin, Chevrot and Poirier20). Additionally, hexarelin and EP80317 are ligands for the CD36 receptor(Reference Febbraio and Silverstein18,Reference Nergiz-Unal, Rademakers and Cosemans19) .
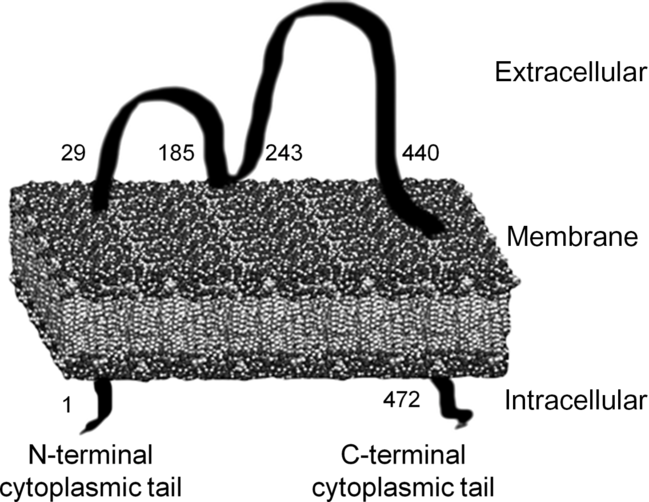
Fig. 1. Transmembrane structure of the cluster of differentiation 36 (CD36) receptor(Reference Nergiz-Unal, Rademakers and Cosemans19,Reference Martin, Chevrot and Poirier20) .
In humans, CD36 is expressed on many cells and tissues including platelets, microvascular endothelial cells, monocytes, macrophages, adipocytes, heart and skeletal muscle cells, retinal pigment epithelial cells and enterocytes, whereas it is partially expressed on hepatocytes and smooth muscle cells(Reference Febbraio and Silverstein18). Initially, CD36 was described as a scavenger receptor that plays a role in endocytosis by binding to its ligands such as TSP1 and oxLDL. Nowadays, CD36 has a variety of metabolic effects since it is expressed in many cells and tissues and has many ligands. Therefore, it is currently being discussed whether CD36 might influence the pathogenesis of hyperlipidaemia, dyslipidaemia, inflammation, atherothrombosis and angiogenesis(Reference Maréchal, Laviolette and Rodrigue-Way17,Reference Febbraio and Silverstein18) .
Fatty acid uptake into cardiomyocytes, adipocytes, enterocytes and skeletal myocytes requires protein-mediated transport by fatty acid transport protein (FATP), fatty acid binding protein (FABP) and CD36(Reference Koonen, Glatz and Bonen21,Reference Glatz and Luiken22) . LCFA uptake into cardiomyocytes is regulated by the vesicular recycling of CD36 from endoplasmic reticulum to the plasma membrane(Reference Glatz and Luiken22). In this context, short-term regulation of LCFA uptake at the plasma membrane is provided both by heart muscle contraction and insulin signalling(Reference Koonen, Glatz and Bonen21). For long-term regulation, expression of CD36 in the nucleus is regulated by PPAR and other transcriptional factors(Reference Glatz, Luiken and Bonen23,Reference Glatz, Nabben and Heather24) . Furthermore, CD36 accelerates the entry of fatty acids from the cytoplasm to mitochondria for β-oxidation(Reference Glatz, Luiken and Bonen23). Since fatty acid uptake into the cytoplasm initiates the signalling pathways related to PPAR, the expressions of genes especially involved in lipid metabolism are circuitously altered by CD36(Reference Glatz, Luiken and Bonen23,Reference Glatz, Nabben and Heather24) . In this context, CD36 may cause the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) and inactivation of sterol regulatory element-binding protein 2 (SREBP2) by inducing PPARγ and thus decrease endogenous cholesterol synthesis(Reference Rodrigue-Way, Caron and Bilodeau16,Reference Demers, Rodrigue-Way and Tremblay25) . Furthermore, CD36-mediated liver X receptor (LXR) activation might accelerate reverse cholesterol transport by up-regulating the activities of ATP-binding cassette transporter (ABC) A1 (ABCA1) and G1 (ABCG1) in the liver and macrophages(Reference Maréchal, Laviolette and Rodrigue-Way17,Reference Bujold, Mellal and Zoccal26,Reference Cuchel and Rader27) . Considering its roles in cholesterol synthesis, transport and reverse cholesterol transport, CD36 might be effective in cholesterol homeostasis and play an important role for dyslipidaemia and hyperlipidaemia-induced CVD(Reference Maréchal, Laviolette and Rodrigue-Way17,Reference Glatz, Luiken and Bonen23) .
In addition to its role for LCFA uptake and lipid metabolism, the binding of CD36 to oxysterols, TSP1, oxLDL and oxidised phospholipids in endothelial cells, macrophages and platelets is important in the onset and development of atherosclerosis by inducing platelet aggregation, thrombosis, production of inflammatory cytokines and foam cell formation(Reference Febbraio and Silverstein18,Reference Nergiz-Unal, Rademakers and Cosemans19,Reference Handberg, Skjelland and Michelsen28,Reference Leonarduzzi, Gargiulo and Gamba29) . Studies have shown that there was a positive correlation between soluble CD36(Reference Handberg, Skjelland and Michelsen28) or macrophage CD36(Reference Nergiz-Ünal, Kuijpers and de Witt30) levels and formation of atherosclerotic plaques. Furthermore, CD36 cooperates with toll-like receptors (TLR) and their heterodimers (TLR4-TLR6) to bind oxLDL and stimulate inflammation-induced atherosclerosis(Reference Sheedy, Grebe and Rayner31). Moreover, it has been reported that CD36 has an important role for the accumulation of cholesterol within the macrophages resulting in lysosomal disruption and activation of NLR family pyrin domain containing 3 (NLRP3) inflammasomes(Reference Sheedy, Grebe and Rayner31,Reference Abumrad and Goldberg32) . Human studies have reported that CD36 increases the release of inflammatory cytokines such as TNFα, IL-1β and interferon-γ by inducing activation of NF-κB in monocyte-derived macrophages(Reference Janabi, Yamashita and Hirano33). On the other hand, CD36 is a receptor for several proteins containing peptide domains known as TSP1 repeats. Thus, in endothelial cells, CD36 functions as an endogenous negative regulator of angiogenesis(Reference Silverstein and Febbraio34). CD36 accomplishes this function by suppressing growth factor-induced pro-angiogenic signals and generating anti-angiogenic signals that cause apoptosis(Reference Silverstein and Febbraio34,Reference Jimenez, Volpert and Crawford35) . All these functions of CD36 reveal the vital role of this receptor in cardiovascular metabolism.
Role of CD36 in cholesterol metabolism
Cholesterol homeostasis is a metabolic process that involves the absorption of dietary cholesterol, endogenous synthesis, transport of cholesterol, cholesterol excretion in the form of bile acids and reabsorption of bile acids. Any change in cholesterol homeostasis might cause pathophysiological conditions and induce increased risk of cardiometabolic disorders(Reference Ahn, Jun and Jang36). The effects of CD36 on lipid metabolism might be different due to its expression level in many cells and the presence of many ligands. As shown in Fig. 2, the possible effects of CD36 on cholesterol homeostasis include absorption of dietary cholesterol, cholesterol synthesis, lipoprotein formation, reverse cholesterol transport, and synthesis and reabsorption of bile acids (Fig. 2)(Reference Maréchal, Laviolette and Rodrigue-Way17,Reference Febbraio and Silverstein18) .
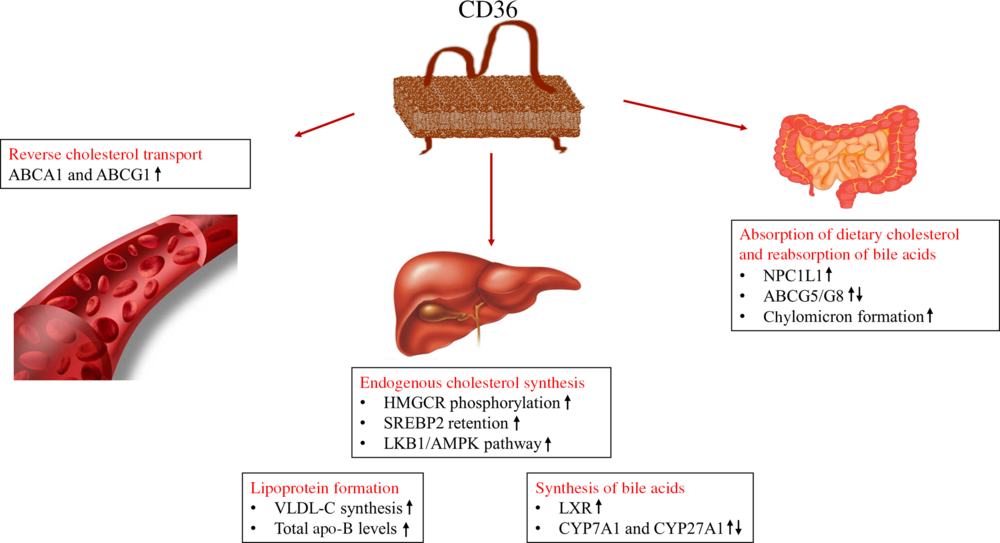
Fig. 2. Roles of cluster of differentiation 36 (CD36) in cholesterol metabolism(Reference Maréchal, Laviolette and Rodrigue-Way17,Reference Febbraio and Silverstein18) . ABC, ATP binding cassette transporters; AMPK, 5′AMP-activated protein kinase; CYP, cytochrome P450 family; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; LKB1, liver kinase B1; LXR, liver X receptor; NPC1L1, Niemann–Pick C1-like 1; SREBP2, sterol regulatory element-binding protein 2; VLDL-C, VLDL-cholesterol.
One of the components of cholesterol homeostasis is the absorption of dietary cholesterol in the gastrointestinal tract. CD36 is expressed in the small intestine, especially on the apical membranes of the duodenum and jejunum, and, therefore, is proposed to facilitate the absorption of fatty acids and cholesterol(Reference Nauli, Nassir and Zheng37). Drugs (i.e. ezetimibe), used in the treatment of hypercholesterolaemia, are known to suppress the absorption of cholesterol in the small intestine by inhibiting important carriers for cholesterol absorption (such as CD36, Niemann–Pick C1-like 1 (NPC1L1), SR-BI)(Reference Ahn, Jun and Jang36,Reference van Bennekum, Werder and Thuahnai38) . In animal studies, it has been shown that dietary cholesterol absorption in the small intestine(Reference Nauli, Nassir and Zheng37,Reference Nassir, Wilson and Han39) and the rate of passage to the lymph circulation of cholesterol(Reference Nauli, Nassir and Zheng37) are lower in CD36 gene knock-out mice. It has also been reported that CD36 increased the absorption of cholesterol by affecting the level of NPC1L1 carrier protein(Reference Nassir, Wilson and Han39). On the other hand, it was found that, in CD36 and SR-BI knock-out mice, cholesterol absorption was delayed with a high-fat diet compared with standard chow feed(Reference Nguyen, Drover and Knopfel40). Additionally, it has been shown that secretion of apo-B to the lymphatic circulation is decreased in CD36 knock-out mice and therefore, the absence of CD36 suppresses chylomicron formation in enterocytes(Reference Nauli, Nassir and Zheng37).
In addition to the effect on the absorption of dietary cholesterol, CD36 might affect cholesterol homeostasis by altering the activities and/or levels of regulatory enzymes for endogenous cholesterol synthesis in many tissues, especially in the liver. CD36 binding to ligands such as hexarelin, oxLDL or oxysterols induces PPARγ expression by activating PPARγ coactivator 1α (PGC1α)(Reference Maréchal, Laviolette and Rodrigue-Way17,Reference Gai, Wang and Visentin41) . PPARγ is a member of a nuclear receptor family that regulates the expression of many glycogenic and lipogenic genes in many tissues, mainly in the liver, cardiomyocytes and adipose tissue(Reference Rodrigue-Way, Caron and Bilodeau16,Reference Maréchal, Laviolette and Rodrigue-Way17,Reference Nicholson and Hajjar42) . Increased expression of PPARγ might decrease endogenous cholesterol synthesis by activating insulin-stimulated gene 1 (Insig1) and insulin-stimulated gene 2 (Insig2) and inducing the degradation of HMGCR and retention of SREBP2 in the endoplasmic reticulum(Reference Maréchal, Laviolette and Rodrigue-Way17). In a study, it was found that expression of PPARγ in adipose tissue was reduced and therefore Insig1 level was decreased in CD36 gene knock-out mice(Reference Hajri, Hall and Jensen43). However, in a recent study, it was reported that CD36 expression increased in macrophages incubated with advanced glycation endproducts, and the expression levels of HMGCR and acetyl-CoA acetyltransferase (ACAT) 1 were elevated(Reference Xu, Wang and Li44). Furthermore, a study conducted on mice showed that the expressions of CD36 and HMGCR were increased in epididymal white adipose tissue, while PPARγ expression was not affected(Reference Lavery, Kurowska-Stolarska and Holmes45). Also, CD36 activates liver kinase B1 (LKB1), which is serine/threonine kinase 11 (STK11), known as a tumour suppressor(Reference Lu, Fang and Huang46). 5′AMP-activated protein kinase α (AMPKα) stimulated via LKB1 activation might inhibit cholesterol synthesis by providing phosphorylation of HMGCR(Reference Maréchal, Laviolette and Rodrigue-Way17,Reference Shackelford and Shaw47) . In a recent study, it was reported that liver CD36 expression was decreased, hepatic fatty acid and cholesterol accumulation was increased, and blood lipoprotein profile was impaired due to decreased activity of the LKB1/AMPK pathway in mice fed with a high-fructose diet(Reference Choi, Abdelmegeed and Song48).
In reverse cholesterol transport, another pathway induced by ligands binding to CD36 is the LXR-mediated pathway, and increased hepatic LXR expression was found to positively correlate with CD36 expression in the liver(Reference Ahn, Jang and Jun49). Increased LXR expression in the liver may have an impact on the levels of ABCA1 and ABCG1 transporters that are important for reverse cholesterol transport, and hence may influence the formation of foam cells in the vessels by altering cholesterol transport from peripheral tissues, primarily from macrophages, to the liver(Reference Maréchal, Laviolette and Rodrigue-Way17,Reference Jessup, Gelissen and Gaus50) . Elevating the level of ABCA1 in macrophages transfers phospholipids and cholesterol to apo-A1, while ABCG1 transfers cholesterol to immature HDL-cholesterol (HDL-C)(Reference Jessup, Gelissen and Gaus50). In CD36 gene knock-out mice, liver expressions of apo-A1 and apo-A4, which are mainly found in the HDL-C structure, were higher than in wild-type mice(Reference Yue, Chen and Nassir51). In addition to reverse cholesterol transport and HDL-C, elevation of CD36 in the liver may induce the synthesis and release of VLDL-cholesterol (VLDL-C) due to the fact that CD36 increases the uptake of LCFA into the liver(Reference Yanai, Chiba and Morimoto52,Reference Yamashita, Hirano and Kuwasako53) . Also, it was found that serum total cholesterol and LDL-cholesterol (LDL-C) levels were significantly elevated in type II CD36-deficient humans (CD36 is expressed on monocytes but not on platelets)(Reference Yamashita, Hirano and Kuwasako53).
Cholesterol is used in the synthesis and reabsorption of bile acids from the intestine, which are metabolic pathways for cholesterol excretion. Increasing levels of CD36 in the liver might cause elevation of the key enzymes of bile acid synthesis, i.e. cholesterol 7α hydroxylase (CYP7A1) and sterol 27 hydroxylase (CYP27A1)(Reference Yuan, Wang and Tian54). Furthermore, studies have reported that CD36-mediated LXR stimulation influences the reabsorption of bile acids by increasing ABCG5/G6 expression and decreasing NPC1L1 transporter expression(Reference Bujold, Mellal and Zoccal26,Reference Ahn, Jang and Jun49) . It was also been reported that an increase in CD36 expression in the hepatocytes of mice fed with a high-fat and high-cholesterol diet leads to an increase in LXR and therefore ABCG5/G8 levels(Reference Kamisako, Tanaka and Kishino55).
Accordingly, ligand-receptor downstream signalling of CD36 might affect cholesterol metabolism via different proposed mechanisms. CD36 may elevate dietary cholesterol absorption, increase or decrease endogenous cholesterol synthesis and reverse cholesterol transport, and may increase synthesis and reabsorption of bile acids(Reference Maréchal, Laviolette and Rodrigue-Way17,Reference Febbraio and Silverstein18,Reference Bujold, Mellal and Zoccal26) . The results of human studies have shown that CD36 deficiency induces dyslipidaemia (elevated blood TAG, total cholesterol and LDL-C, and decreased HDL-C) and increases the concentration of lipoprotein remnants(Reference Yanai, Chiba and Morimoto52,Reference Yamashita, Hirano and Kuwasako53) . However, more preclinical and clinical studies are needed to clarify the mechanisms of how CD36 affects cholesterol metabolism.
Dietary fatty acids, CD36 and cholesterol metabolism
The dietary fatty acid pattern is essential for cardiovascular health which may increase or decrease blood total cholesterol, and circulating lipoprotein and apolipoprotein levels(5,Reference Eckel, Jakicic and Ard7,56) . Moreover, the saturation (saturated, monounsaturated, polyunsaturated) and the form (cis/trans) of fatty acids may change the expression of CD36 in many tissues and cells involved in cholesterol homeostasis, especially in the liver, small intestine, adipose tissue and macrophages(Reference Nigro, Menotti and Cento12,Reference Nergiz-Ünal, Kuijpers and de Witt30,Reference Kamisah, Periyah and Lee57) . Therefore, the studies on the combining effects of novel biomarkers (i.e. CD36) and dietary fatty acid intake on cholesterol metabolism have been currently accumulating in the literature.
SFA and CD36-mediated cholesterol metabolism
Dietary SFA might have an impact on cholesterol homeostasis due to various mechanisms including absorption of dietary cholesterol in the small intestine, endogenous cholesterol synthesis, circulating lipoproteins and cholesterol excretion in the form of bile acids(Reference Ooi, Watts and Ng6,Reference de Wit, Derrien and Bosch-Vermeulen58) . Depending on the source of food, high dietary SFA intake resulted in higher serum total cholesterol, LDL-C(Reference Sun, Neelakantan and Wu59,Reference Mozaffarian, Micha and Wallace60) and non-HDL-C levels(Reference Key, Appleby and Bradbury61) in meta-analyses. It was also shown that consumption of red and processed meat was positively associated with serum non-HDL-C level(Reference Key, Appleby and Bradbury61). Though, since dairy products, butter, red meat, fish, eggs and poultry have different SFA contents(56), they might influence cholesterol homeostasis depending on the dietary fatty acid pattern(Reference Key, Appleby and Bradbury61,Reference de Goede, Geleijnse and Ding62) .
Cholesterol absorption
NPC1L1 has been identified as a cholesterol transporter localised at the apical membrane of the small intestine; while ABCA1, ABCG5 and ABCG8 are presumed to facilitate cholesterol efflux from the enterocyte(Reference Jia, Betters and Yu63). As summarised in Table 1, it has been generally hypothesised that high dietary SFA might result in elevated cholesterol absorption (NPC1L1 and CD36), chylomicron formation (microsomal TAG transfer protein (MTTP) and ACAT), and cholesterol efflux from enterocytes to lymphatic circulation (ABCA1/G1/G5/G8)(Reference de Vogel-van den Bosch, de Wit and Hooiveld64-Reference Zhu-qin, Hou-zao and Rui-feng68). These results showed that the absorption/transport of cholesterol from the apical and basolateral membranes of enterocytes was a complex process with many carriers, proteins and enzymes. In this context, the hypothesis of acceleration of cholesterol absorption and chylomicron formation in the small intestine via CD36 has been presented in the literature in the last decade(Reference Nauli, Nassir and Zheng37,Reference Nassir, Wilson and Han39) . Expression of CD36 at the proximal end of the small intestine was higher than at the distal end of the body and cholesterol absorption decreased by 50 % in CD36(–/–) mice compared with CD36(+/+) mice(Reference Nauli, Nassir and Zheng37). Hence, down-regulation, dysfunction or deficiency of CD36 in the intestine might lead to a delay of efficient chylomicron formation (MTTP, FABP) and clearance (apo-C2) in the jejunum induced by impaired gene expression(Reference Nauli, Nassir and Zheng37,Reference Nassir, Wilson and Han39) . A study conducted on mice fed with a high-SFA diet showed that although CD36 and NPC1L1 expressions in the small intestine did not change, the expressions of diacylglycerol acyltransferase 1 (DGAT1), MTTP, FABP and apo-B increased in enterocytes, inducing chylomicron formation(Reference Buttet, Poirier and Traynard14).
Table 1. Potential effects of SFA on CD36-related cholesterol homeostasis
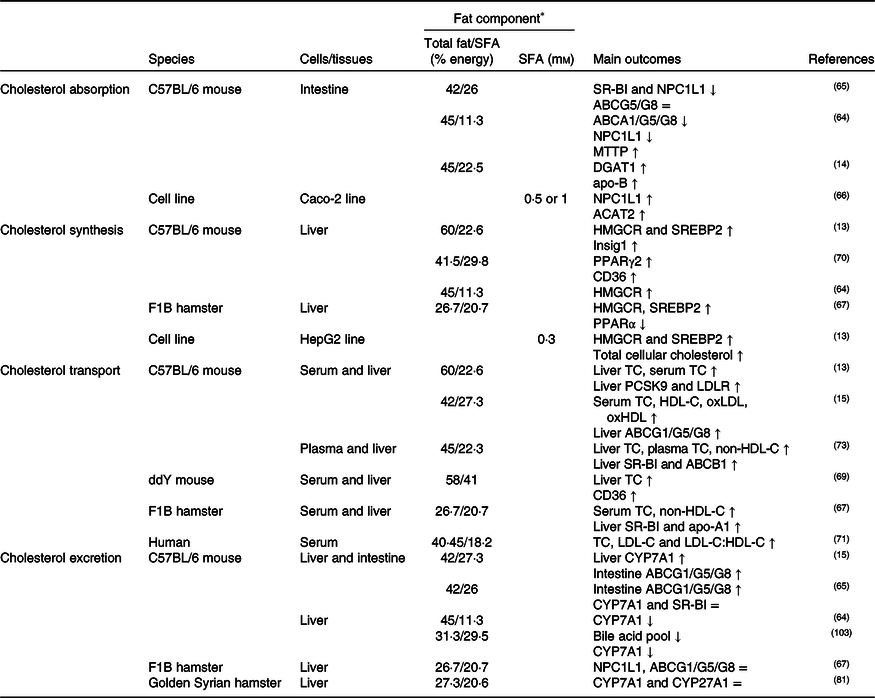
↑, Increase; ↓, decrease; =, no change; ABC, ATP binding cassette transporter; ACAT, acetyl-CoA acetyltransferase; CD36, cluster of differentiation 36; CYP7A1, cholesterol 7α hydroxylase; CYP27A1, sterol 27 hydroxylase; DGAT1, diacylglycerol acyltransferase 1; HDL-C, HDL-cholesterol; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; Insig1, insulin-stimulated gene 1; LDL-C, LDL-cholesterol; LDLR, LDL receptor; MTTP, microsomal TAG transfer protein; NPC1L1, Niemann–Pick C1-like 1; oxHDL, oxidised HDL; oxLDL, oxidised LDL; PCSK9, proprotein convertase subtilisin/kexin type 9; SR-BI, scavenger receptor class B type I; SREBP2, sterol regulatory element-binding protein 2; TC, total cholesterol.
* The molarity or percentage of the energy for total fat/SFA is recorded or calculated from the diets in the reference studies.
Cholesterol synthesis
SFA might induce the endogenous synthesis of cholesterol in many tissues. As shown in Table 1, in this process, SREBP2 might be up-regulated by dietary SFA which regulates the synthesis and uptake of cholesterol by altering the expression of HMGCR and LDL receptor (LDLR)(Reference Nigro, Menotti and Cento12,Reference Lecker, Matthan and Billheimer67) . Additionally, PPARγ coactivator 1α (PGC1α) may be stimulated by dietary SFA and contribute to higher SREBP2 mRNA levels. In the endoplasmic reticulum, a sterol sensor was proposed for the modulation of SREBP2 transcriptional activity in response to changes in intracellular free cholesterol levels(Reference Lecker, Matthan and Billheimer67). Although ACAT2 is responsible for the esterification of cholesterol, the effects of high SFA intake on ACAT2 have not been clarified yet(Reference Zhu-qin, Hou-zao and Rui-feng68). Mice fed with a high-SFA diet showed increased endogenous cholesterol synthesis by decreasing expression of CD36 and thus expression of PPARγ in the liver(Reference Yamazaki, Kishimoto and Miura69,Reference Yamazaki, Shiraishi and Kishimoto70) . SFA are generally thought to increase endogenous cholesterol synthesis, but the effect of CD36 on this mechanism is still unclear.
Cholesterol transport
Intervention studies showed that SFA elevated atherogenic lipoproteins/apolipoproteins and decreased non-atherogenic lipoproteins/apolipoproteins(Reference Lamarche and Couture11,Reference Sun, Neelakantan and Wu59,Reference Kien, Bunn and Stevens71-Reference Tholstrup, Hjerpsted and Raff75) as summarised in Table 1. It was also reported that high dietary SFA intake increased the oxidation of lipoproteins (such as oxLDL and oxHDL) in serum compared with dietary unsaturated fatty acids(Reference Cedo, Metso and Santos15). In the literature, it has been generally reported that high dietary SFA intake might decrease plasma HDL-C levels. However, a meta-analysis found that high dietary SFA intake from hydrogenated vegetable oils consisting of TFA might elevate plasma HDL-C(Reference Sun, Neelakantan and Wu59). Likewise, high dietary SFA due to dietary total fat intake might cause higher plasma HDL-C level relative to increase in plasma total cholesterol level in the case of hyperlipidaemia(Reference Schwingshackl and Hoffmann76). In addition, cholesteryl ester transfer protein (CETP) might be up-regulated by high SFA intake, induce cholesterol transport from HDL-C to VLDL-C, and thus decrease reverse cholesterol transport(Reference De Vries, Beusekamp and Kerstens77). Moreover, high dietary SFA has been shown to up-regulate(Reference Cedo, Metso and Santos15,Reference Wang, Matthan and Wu78) , down-regulate(Reference Wu, Sarna and Hwang13) or not change(Reference Miles, Wallace and Calder79) the expression of transporters such as CD36, SR-BI, ABCA1 and ABCG1 in reverse cholesterol transport in the liver and macrophages.
Cholesterol excretion
As shown in Table 1, high dietary SFA might inhibit the synthesis of bile acids in the liver via CYP7A1 and CYP27A1 enzymes, and might accelerate the reabsorption of bile acids via ABCG5/G8 in the intestine(Reference Cedo, Metso and Santos15,Reference de Vogel-van den Bosch, de Wit and Hooiveld64,Reference Jung, Millman and Tall80,Reference Loison, Mendy and Serougne81) . Moreover, activation of LXR increases bile acid synthesis from cholesterol and cholesterol excretion into bile. CD36 might be involved in this mechanism due to the fact that it stimulates LXR expression(Reference Maréchal, Laviolette and Rodrigue-Way17,Reference Ahn, Jang and Jun49) . However, studies on how SFA affect CD36-mediated bile acid synthesis in the liver and reabsorption in the intestine are not completely clarified yet.
To sum up, high dietary SFA intake might impair cholesterol homeostasis by a variety of mechanisms including absorption, synthesis, transport and excretion of cholesterol (Table 1). In these mechanisms, many enzymes, transcription factors and transporters such as CD36 may be influenced by dietary SFA content. Thus, according to the results of recent studies it was indicated that SFA of 12–43 % from daily energy intake disrupts cholesterol homeostasis and elevates atherogenic lipoprotein levels in the blood(Reference Sun, Neelakantan and Wu59,Reference Kien, Bunn and Stevens71,Reference Raz, Steinvil and Berliner74) . International authorities recommend limiting dietary SFA due to the adverse effects of SFA intake on cholesterol metabolism and homeostasis(5,Reference Eckel, Jakicic and Ard7,Reference Siri-Tarino and Krauss82) . In this context, the European Food Safety Authority (EFSA) recommends reducing SFA intake as low as possible in order to prevent the elevation of LDL-C(83), while the American Heart Association (AHA) recommends limiting SFA to 5–6 % of the daily energy intake for healthy adults(Reference Eckel, Jakicic and Ard7). The European Society of Cardiology, the 2015–2020 US Department of Agriculture Dietary Guidelines for Americans, and Turkey Dietary Guidelines(84) recommend that dietary SFA should be <10 % (as little as possible) of daily energy intake(Reference Perk, De Backer and Gohlke1,84) . Therefore, since dietary SFA might influence CD36-induced cholesterol homeostasis, the importance of CD36 in human nutrition and diet therapy should be considered in the future.
MUFA and CD36-mediated cholesterol metabolism
Cholesterol absorption
Intestinal cholesterol absorption and chylomicron formation involve several cholesterol transporters including NPC1L1, ABCG5/8, SR-BI and CD36(Reference Hui and Howles85), enzymes and proteins such as apo-B48, MTTP and and ACAT2(Reference Abumrad and Davidson86). Since absorption of dietary cholesterol mainly occurs in the proximal side of the small intestine, NPC1L1, CD36 and ABCG5/G8 are highly expressed on the proximal side(Reference Sontag, Chellan and Getz87). According to results of the studies, a high MUFA intake might decrease cholesterol absorption by suppressing the expression of these transporters(Reference Ooi, Watts and Ng6,Reference Eckel, Jakicic and Ard7,Reference Chen, Li and Zhang88,Reference Goncalves, Gleize and Roi89) or might not affect the absorption (Table 2)(Reference Yang, Chen and Ma66,Reference Lecker, Matthan and Billheimer67,Reference Chen, Li and Zhang88) . It was also reported that CD36 expression in enterocytes increased after high olive oil intake in mice(Reference Sontag, Chellan and Getz87). Although this suggests that a high MUFA intake might increase cholesterol absorption, the increase in cholesterol absorption might be due to high fat intake rather than high MUFA intake.
Table 2. Potential effects of MUFA on CD36-related cholesterol homeostasis
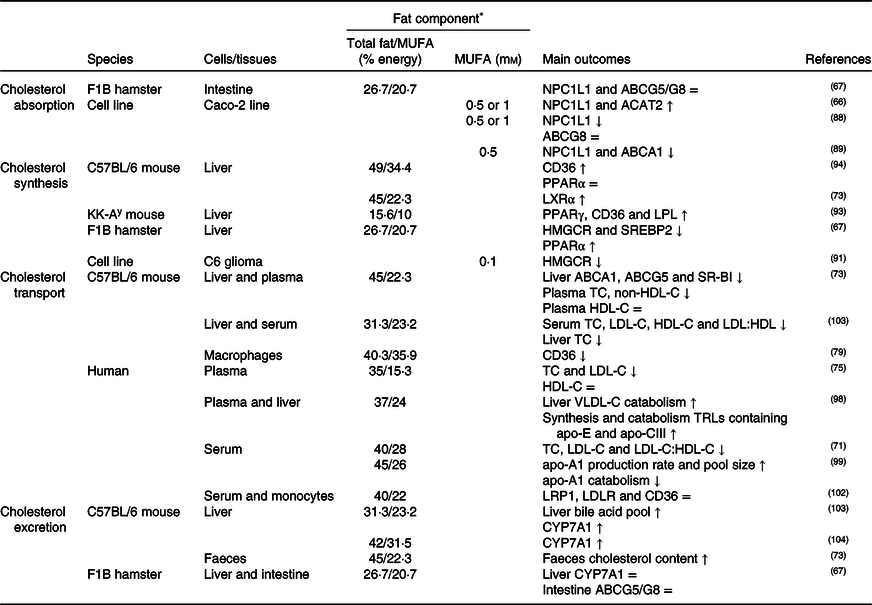
↑, Increase; ↓, decrease; =, no change; ABC, ATP binding cassette transporter; ACAT, acetyl-CoA acetyltransferase; CD36, cluster of differentiation 36; CYP7A1, cholesterol 7α hydroxylase; HDL-C, HDL-cholesterol; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; LDL-C, LDL-cholesterol; LDLR, LDL receptor; LPL, lipoprotein lipase; LRP1, LDL receptor-related protein-1; LXR, liver X receptor; NPC1L1, Niemann–Pick C1-like 1; SR-BI, scavenger receptor class B type I; SREBP2, sterol regulatory element-binding protein 2; TC, total cholesterol; TRLs, TAG rich lipoproteins; VLDL-C, VLDL-cholesterol.
* The molarity or percentage of the energy for total fat/MUFA is recorded or calculated from the diets in the reference studies.
Cholesterol synthesis
As summarised in Table 2, the MUFA content of the diet might decrease endogenous cholesterol synthesis by reducing HMGCR and SREBP2 expression(Reference Lecker, Matthan and Billheimer67,Reference Agarwal-Mawal, Murray and Belkhode90,Reference Natali, Siculella and Salvati91) . Although these effects are not clear, studies have shown that a high oleic acid intake reduces HMGCR activity(Reference Agarwal-Mawal, Murray and Belkhode90) or does not have an impact on HMGCR enzyme level(Reference Natali, Siculella and Salvati91). A high consumption of MUFA may induce LDLR expression by stimulating ACAT1 and by increasing intracellular cholesteryl ester content(Reference Goldstein, DeBose-Boyd and Brown92). In addition, CD36 stimulates PPARγ expression in the liver, and therefore might suppress cholesterol synthesis. In rodent studies, it has been reported that a high consumption of MUFA elevates CD36 and PPARγ expression, and accordingly decreases plasma and liver total cholesterol levels(Reference Yang, Miyahara and Iwasaki93,Reference Jurado-Ruiz, Varela and Luque94) . These results of studies propose that a relatively higher dietary MUFA content might reduce cholesterol biosynthesis and the levels of plasma and liver total cholesterol to prevent dyslipidaemia or hyperlipidaemia.
Cholesterol transport
MUFA are generally thought to have positive effects on cholesterol homeostasis(Reference Jebb, Lovegrove and Griffin95,Reference Mozaffarian and Clarke96) . The results of studies reported that high dietary MUFA intake might reduce atherogenic lipoproteins/apolipoproteins(Reference Lamantia, Sniderman and Faraj72,Reference Peou, Milliard-Hasting and Shah97,Reference Zheng, Khoo and Furtado98) , and might increase non-atherogenic lipoproteins/apolipoproteins(Reference Kien, Bunn and Stevens71,Reference Labonte, Jenkins and Lewis99) , as shown in Table 2. In contrast, as high dietary MUFA intake might result in high dietary fat intake, the levels of total cholesterol, LDL-C and non-HDL-C might increase after high MUFA intake(Reference O’Reilly, Dillon and Guo73). Moreover, high dietary MUFA might activate the synthesis of TAG-rich lipoproteins containing apo-E and apo-CIII and therefore might accelerate the catabolic pathways for these TAG-rich lipoproteins(Reference Zheng, Khoo and Furtado98). In macrophages, since CD36-mediated PPARγ and LXR activation may suppress ABCA1 and ABCG1 expression, decreasing ABCA1/G1 levels might cause impairment of reverse cholesterol transport(Reference Jiang and Li100). In human studies, it has been found that the CD36 in monocytes is decreased(Reference Valderas-Martinez, Chiva-Blanch and Casas101) or is not affected(Reference Llorente-Cortes, Estruch and Mena102) in the individuals consuming a high intake of olive oil. Studies on how MUFA influence the CD36-mediated circulating lipoproteins levels and reverse cholesterol metabolism are controversial depending on the fatty acid source of the food, other dietary components, research model and dietary manipulation.
Cholesterol excretion
Removal of excess cholesterol from the body in the form of bile acids in the liver is one of the important steps for maintaining cholesterol homeostasis(Reference Li, Hou and Ma103). It has been reported that a high MUFA intake might up-regulate the synthesis of bile acids in the liver(Reference Li, Hou and Ma103,Reference Yang, Pryor and Noguchi104) , but might have no significant effect on the expression of transporters involving the reabsorption of bile acids (Table 2)(Reference Lecker, Matthan and Billheimer67,Reference Goncalves, Gleize and Roi89) . The mechanisms of synthesis and release of bile acids in the liver and the reabsorption of bile acids in the intestine might be affected by the CD36 receptor in the liver and intestines(Reference Yuan, Wang and Tian54,Reference Kamisako, Tanaka and Kishino55) . Studies have reported that a diet high in MUFA up-regulates the expression of CD36 in the liver(Reference Jurado-Ruiz, Varela and Luque94) and small intestine(Reference Nassir, Wilson and Han39). However, because the effects of dietary MUFA on cholesterol excretion are not yet clear, there is need for further randomised, controlled human studies related to the effect of high MUFA intake on CD36, and other related factors in bile metabolism.
Basically, it is accepted that sufficient dietary MUFA improves cholesterol homeostasis and prevents the impairment of cholesterol metabolism. Studies have shown that 15–25 % of total energy from MUFA decreases the endogenous synthesis of cholesterol(Reference Lecker, Matthan and Billheimer67,Reference Agarwal-Mawal, Murray and Belkhode90) and levels of atherogenic lipoproteins/apoproteins, but increases non-atherogenic lipoproteins/apoproteins(Reference O’Reilly, Dillon and Guo73,Reference Labonte, Jenkins and Lewis99) and cholesterol excretion via increased synthesis of bile acids(Reference Li, Hou and Ma103). Therefore, the guidelines published in European countries recommend that daily MUFA intake to be 10–15 % of the total energy intake for healthy adults(Reference Becker, Lyhne and Pedersen105). Also, the AHA(106) and Turkey Dietary Guidelines(84) recommend increasing dietary MUFA intake, while decreasing dietary SFA(84,106) . Nonetheless, there is need for more preclinical and clinical studies to combine the effects of dietary MUFA and CD36-mediated cholesterol homeostasis.
Trans-fatty acids and CD36-mediated cholesterol metabolism
Dietary TFA are generally assumed to have adverse effects on cholesterol metabolism(Reference Ooi, Watts and Ng6,Reference Gebauer, Chardigny and Jakobsen107) . TFA might be formed industrially in the production process of foods as well as naturally in animal-based foods(Reference Gebauer, Destaillats and Dionisi9,Reference Laake, Pedersen and Selmer108) . Although the effects of TFA both obtained from animal-based foods(Reference Gebauer, Destaillats and Dionisi9) and processed foods(Reference Gebauer, Destaillats and Dionisi9,Reference Mori, Ishida and Yasuda109) on CD36-mediated cholesterol metabolism have not been explained yet, the general belief is that they may disrupt cholesterol homeostasis.
High dietary TFA intake impairs the blood lipoprotein profile by elevating the levels of blood total cholesterol(Reference Gebauer, Destaillats and Dionisi9,Reference Neuschwander-Tetri, Ford and Acharya110) , VLDL-C(Reference Afonso, Lavrador and Koike111), LDL-C, apo-B(Reference Bendsen, Chabanova and Thomsen112) and Lp(a)(Reference Gebauer, Destaillats and Dionisi9) and by decreasing HDL-C and apo-A1 levels(Reference Lamantia, Sniderman and Faraj72,Reference Afonso, Lavrador and Koike111) . However, the consumed amount of TFA is important and ≤0·6 % of the daily energy requirement has no significant effect on cholesterol metabolism(Reference Takeuchi, Yamaki and Hirose113). In parallel, a few epidemiological(Reference Gebauer, Chardigny and Jakobsen107) and randomised controlled studies(Reference Gayet-Boyer, Tenenhaus-Aziza and Prunet114) have reported that TFA (2–4 % of daily energy) taken from animal-based foods had no significant effect on blood lipoprotein profile and would not pose a risk for CVD. On the other hand, a study concluded that replacement of 1 % of the daily energy intake of TFA with SFA, MUFA or PUFA decreased the ratios of total cholesterol:HDL-C and apo-B:apo-A1 and Lp(a) levels(Reference Mozaffarian and Clarke96).
There are few studies in the literature on the effects of TFA on CD36-mediated cholesterol metabolism. In a study conducted on mice, it was reported that high dietary TFA intake resulted in an increased level of CD36 in hepatocytes and thus induced the synthesis and release of apo-B and VLDL-C(Reference Li, Viswanadha and Loor115). On the contrary, it was been reported that a diet high in TFA did not alter CD36 expression in hepatocytes and cardiomyocytes in mice(Reference Ganguly, LaVallee and Maddaford116). Nevertheless, it is not clear how TFA affect CD36-mediated cholesterol metabolism in the liver and other tissues involved in cholesterol homeostasis.
The literature has indicated that taking more than 0·6 % of total energy from TFA has a negative impact on cholesterol homeostasis(Reference Mozaffarian and Clarke96,Reference Takeuchi, Yamaki and Hirose113) , while the potential factors affecting these mechanisms such as CD36 need to be further explained. Due to the undesirable effects of TFA on cholesterol metabolism, EFSA recommends the reduction of TFA intake as much as possible(5). The Turkey Dietary Guidelines and AHA reported that daily TFA intake should be less than 1 % of total energy(Reference Eckel, Jakicic and Ard7,84) . In this context, more studies are needed to combine the effects of TFA on cholesterol homeostasis mediated by CD36 and other related factors.
PUFA and CD36-mediated cholesterol metabolism
Dietary n-3 PUFA and n-6 PUFA amounts and n-3:n-6 PUFA ratio are important for cholesterol homeostasis, since the amount and pattern of PUFA in the diet may distinctly affect cholesterol metabolism(Reference Ooi, Watts and Ng6,Reference Eckel, Jakicic and Ard7) . Thus, the dietary PUFA pattern might influence several mechanisms related to cholesterol homeostasis including the absorption, endogenous synthesis and transport of cholesterol, and bile acid metabolism(Reference Yuan, Wang and Tian54). Although PUFA generally have positive effects on the metabolism, the effects of excess intake of n-6 PUFA on cholesterol homeostasis are debatable. Hence, international guidelines for nutrition recommend paying attention to the consumed amounts of PUFA(5,Reference Eckel, Jakicic and Ard7,Reference Mozaffarian and Ludwing117) . Therefore, for healthy adults, EFSA recommends taking 5–10 % of daily energy intake from PUFA and increasing the n-3 PUFA:n-6 PUFA ratio(5); the Turkey Dietary Guidelines and AHA recommend that PUFA intake should be 7–10 % of daily total energy intake(Reference Eckel, Jakicic and Ard7,84) .
n-3 PUFA and CD36-mediated cholesterol metabolism
Cholesterol absorption
Dietary cholesterol is absorbed from micelles with fatty acids and phospholipids in the proximal parts of the small intestine, re-esterified into cholesteryl esters for the assembly into lipoproteins, and transported to the lymph and then to the circulation(Reference Yang, Chen and Ma66). As shown in Table 3, studies have shown that n-3 PUFA inhibit cholesterol uptake and transport by down-regulating the expression levels of NPC1L1 and ABCA1/G5(Reference Yang, Chen and Ma66,Reference Goncalves, Gleize and Roi89,Reference Chen, Jiang and Liang118) . Moreover, caveolin 1, as a chaperone complex, regulates cholesterol influx or efflux via plasma membrane caveolae. A cell culture study reported that cholesterol absorption might be inhibited by down-regulating caveolin 1 expression in Caco-2 cells incubated with n-3 PUFA(Reference Yang, Chen and Ma66). The combined effects of NPC1L1, ABC transporters, SR-BI and CD36 may play a critical role in modulating the amount of cholesterol that eventually reaches the lymph from the intestinal lumen(Reference Yang, Chen and Ma66,Reference Goncalves, Gleize and Roi89) .
Table 3. Potential effects of n-3 PUFA on CD36-related cholesterol homeostasis
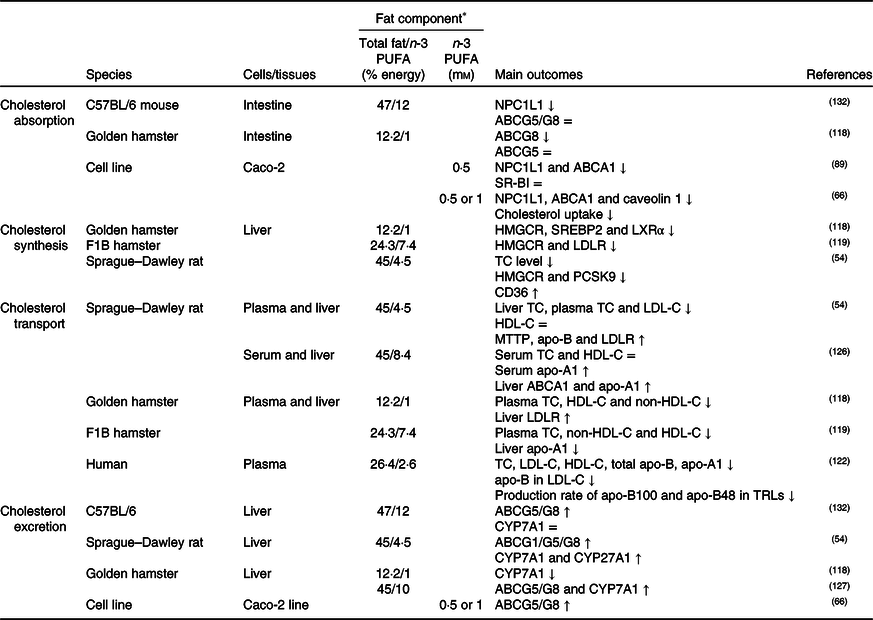
↑, Increase; ↓, decrease; =, no change; ABC, ATP binding cassette transporter; CD36, cluster of differentiation 36; CYP7A1, cholesterol 7α hydroxylase; CYP27A1, sterol 27 hydroxylase; HDL-C, HDL-cholesterol; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; LDL-C, LDL-cholesterol; LDLR, LDL receptor; LXR, liver X receptor; MTTP, microsomal TAG transfer protein; NPC1L1, Niemann–Pick C1-like 1; PCSK9, proprotein convertase subtilisin/kexin type 9; SR-BI, scavenger receptor class B type I; SREBP2, sterol regulatory element-binding protein 2; TC, total cholesterol; TRLs, TAG-rich lipoproteins.
* The molarity or percentage of the energy for total fat/n-3 PUFA is recorded or calculated from the diets in the reference studies.
Cholesterol synthesis
It was generally thought that consumption of fish oil (source of n-3 PUFA) might suppress cholesterol synthesis by inhibiting HMGCR, SREBP2, proprotein convertase subtilisin/kexin type 9 (PCSK9) and LXR expression and by up-regulating CD36 expression in the liver(Reference Yang, Chen and Ma66,Reference Lecker, Matthan and Billheimer67,Reference Wang, Matthan and Wu78,Reference Chen, Jiang and Liang118,Reference Lecker, Matthan and Billheimer119) , as summarised in Table 3. As a result of decreasing cholesterol synthesis, it is supposed that liver cholesterol level, especially cholesteryl ester rather than free cholesterol, decreases(Reference Wang, Matthan and Wu78,Reference Park, Choi and Jin120) . In this context, it has been reported that ACAT1 expression is elevated in parallel with decreased CD36 expression in the aorta of apo-E knock-out mice fed with a diet high in EPA and DHA(Reference Liu, Hu and Wu121). Evaluating the potential mechanisms related to the effects of n-3 PUFA on CD36-induced cholesterol absorption and biosynthesis, n-3 PUFA might suppress dietary cholesterol absorption and cholesterol synthesis- and metabolism-associated mechanisms.
Cholesterol transport
As shown in Table 3, high dietary n-3 PUFA intake might decrease atherogenic lipoproteins/apolipoproteins(Reference Lamantia, Sniderman and Faraj72,Reference Ooi, Lichtenstein and Millar122,Reference Vidakovic, Jaddoe and Voortman123) and oxidised lipoproteins (i.e. oxLDL, oxHDL)(Reference Chang, Seo and Du124,Reference Yang, Song and Yin125) , but might increase anti-atherogenic lipoproteins/apolipoproteins in the blood(Reference Lamantia, Sniderman and Faraj72,Reference Vidakovic, Jaddoe and Voortman123,Reference Xie, Zhang and Zhao126) . In reverse cholesterol transport, hepatic HDL-C uptake is one of the important steps involving enzymes and transporters such as ABCA1/G1, SR-BI and lecithin cholesterol acyltransferase (LCAT), and an increased amount of n-3 PUFA intake might accelerate HDL-C uptake by increasing the expression of these proteins(Reference Xie, Zhang and Zhao126,Reference Kasbi Chadli, Nazih and Krempf127) . Additionally, CD36 in macrophages might involve reverse cholesterol transport. In this context, it has been reported that n-3 PUFA intake up-regulates CD36 expression in addition to ABCA1/G1 in macrophages and hepatocytes and therefore accelerates reverse cholesterol transport(Reference McLaren, Michael and Guschina128,Reference Starcevic, Filipovic and Galan129) . On contrary, it has been shown that n-3 PUFA do not alter(Reference Nergiz-Ünal, Kuijpers and de Witt30,Reference Wang, Wu and Matthan130) or decrease(Reference Liu, Hu and Wu121) CD36 expression in macrophages(Reference Nergiz-Ünal, Kuijpers and de Witt30,Reference Wang, Wu and Matthan130) . Moreover, up-regulation of CD36 expression might cause the degradation of PCSK9(Reference Yuan, Wang and Tian54), which has a role in the degradation of the LDLR, and therefore down-regulation of PCSK9 might result in a decrease in LDLR degradation and increase circulating LDL-C levels(Reference Blanchard, Khantalin and Ramin-Mangata131). As pointed out in the indicated studies, roles of CD36 in oxidised lipid uptake as a scavenger receptor and in fatty acid uptake as a fatty acid transporter in reverse cholesterol transport have been intensively studied. However, the literature is still gathering on the effects of dietary n-3 PUFA and CD36-mediated cholesterol transport and lipoprotein homeostasis.
Cholesterol excretion
Cholesterol excretion via bile acids might be induced by high dietary n-3 PUFA by elevating the expression of CD36, CYP7A1 and CYP27A1 in the liver and ABCG1/G5/G8 transporters in the liver and intestine (Table 3)(Reference Yuan, Wang and Tian54,Reference Yang, Chen and Ma66,Reference Park, Choi and Jin120,Reference Kasbi Chadli, Nazih and Krempf127,Reference Nishimoto, Pellizzon and Aihara132) . In contrast, a study has reported that the liver expression of CYP7A1 in hamsters fed with high DHA is reduced. The study suggested that it might depend on low liver total cholesterol levels due to low HMGCR expression(Reference Chen, Jiang and Liang118). Yet, there is limited knowledge about the dietary n-3 PUFA associated with CD36-induced bile acid metabolism.
Considering the proposed mechanisms in the literature, high dietary n-3 PUFA (in the form of fish oil, EPA or DHA) and/or n-3:n-6 PUFA ratio (1:3–1:10) might suppress cholesterol absorption and endogenous synthesis, and accelerate reverse cholesterol transport, secretion of bile acids and cholesterol excretion. With these potential mechanisms, CD36 is a substantial factor and more studies are needed to elucidate the mechanisms including n-3 PUFA and CD36-signalling pathways.
n-6 PUFA and CD36-mediated cholesterol metabolism
Cholesterol absorption
The effects of n-6 PUFA on dietary cholesterol homeostasis still need to be clarified because n-6 PUFA might improve or impair cholesterol homeostasis with different metabolic processes involved in absorption, synthesis, transport and excretion of cholesterol(Reference González-Becerra, Ramos-Lopez and Barrón-Cabrera133). An in vitro study reported that Caco-2 cells incubated with linoleic acid and arachidonic acid did not affect the level of NPC1L1(Reference Yang, Chen and Ma66,Reference Goncalves, Gleize and Roi89) and SR-BI(Reference Goncalves, Gleize and Roi89), whereas arachidonic acid decreased ABCA1 expression (Table 4)(Reference Yang, Chen and Ma66,Reference Goncalves, Gleize and Roi89) . Nevertheless, there is limited knowledge about the effects of dietary n-6 PUFA on cholesterol absorption in the intestine.
Table 4. Potential effects of n-6 PUFA on CD36-related cholesterol homeostasis
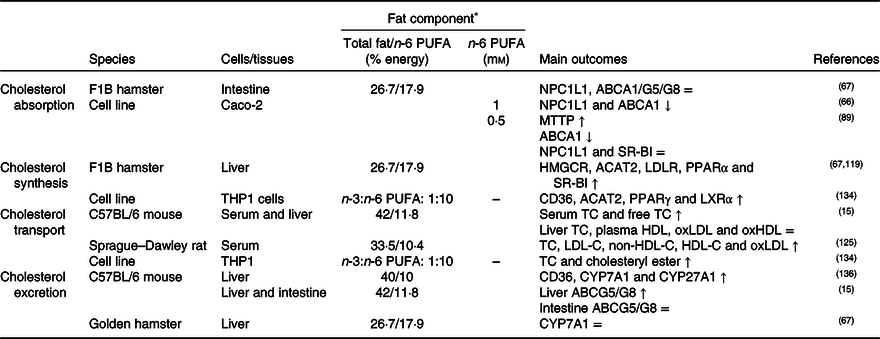
↑, Increase; ↓, decrease; =, no change; ABC, ATP binding cassette transporter; ACAT, acetyl-CoA acetyltransferase; CD36, cluster of differentiation 36; CYP7A1, cholesterol 7α hydroxylase; CYP27A1, sterol 27 hydroxylase; HDL-C, HDL-cholesterol; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; LDL-C, LDL-cholesterol; LDLR, LDL receptor; LXR, liver X receptor; MTTP, microsomal TAG transfer protein; NPC1L1, Niemann–Pick C1-like 1; oxHDL, oxidised HDL; oxLDL, oxidised LDL; SR-BI, scavenger receptor class B type I; TC, total cholesterol.
* The molarity or percentage of the energy for total fat/n-6 PUFA is recorded or calculated from the diets in the reference studies.
Cholesterol synthesis
CD36 might induce PPARγ and therefore inhibit cholesterol biosynthesis. A decrease in the n-3:n-6 PUFA ratio of the diet (1:10) may increase the expression of CD36, PPARγ, LXR and ACAT1 in macrophages(Reference Song, Xia and Yang134). In literature, it was concluded that CD36 expression was up-regulated by dietary sunflower-seed oil in mice and suppressed the endogenous synthesis of cholesterol(Reference Yamazaki, Kishimoto and Miura69). However, high dietary n-6 PUFA might decrease SREBP2 expression(Reference Yang, Chen and Ma66) and might not have a significant effect on HMGCR enzyme(Reference Lecker, Matthan and Billheimer119). n-6 PUFA might also induce cholesterol accumulation in the liver by elevating the level of ACAT2 and esterification of cholesterol in the hepatocytes (Table 4)(Reference Lecker, Matthan and Billheimer119).
Cholesterol transport
As a result of studies investigating the effects of high dietary n-6 PUFA on the lipoprotein profile, n-6 PUFA might elevate blood LDL-C, VLDL-C(Reference Ooi, Watts and Ng6,Reference Lamantia, Sniderman and Faraj72,Reference Choo, Ueshima and Curb135) , HDL-C and apo-A1 levels and might decrease apo-B by accelerating its catabolism(Reference Ooi, Watts and Ng6,Reference Lamantia, Sniderman and Faraj72) . An increase in n-6 PUFA in the diet may increase plasma oxLDL(Reference Yang, Song and Yin125), but decrease the formation of oxLDL and oxHDL compared with a high SFA intake(Reference Cedo, Metso and Santos15). Furthermore, it has been reported that a dietary n-3:n-6 PUFA ratio of 1:20 increases the levels of reactive oxygen species and oxLDL in the blood(Reference Yang, Song and Yin125), but that the n-3:n-6 ratio of higher than 1:5 decreases the total cholesterol content in macrophages in atherosclerosis (Table 4)(Reference Song, Xia and Yang134).
Cholesterol excretion
In cholesterol excretion in the form of bile acids, it has been pointed out that consumption of high amounts of soyabean oil elevate the expression of CD36 and decrease the expression of bile acid synthesis-related enzymes such as CYP7A1 and CYP27A1 in mouse liver(Reference Deol, Evans and Dhahbi136). On the other hand, linoleic acid and arachidonic acid did not affect the expression of ABCG5 and ABCG8, transporters related to excretion and reabsorption of cholesterol in the form of bile acids, in the small intestine (Table 4)(Reference Yang, Chen and Ma66).
It was been suggested that both n-6 PUFA amount and the n-3:n-6 ratio of the diet affect CD36 and other endogenous factors related to cholesterol homeostasis. However, studies on how n-6 PUFA influence CD36-mediated cholesterol homeostasis are limited in the literature and, therefore, the influences of n-6 PUFA intake on cholesterol metabolism with underlying mechanisms should be clarified.
Conclusion
Hypercholesterolaemia and dyslipidaemia, which are CVD risk factors, are known to be influenced by dietary fat and the fatty acid pattern determined in the current literature. As part of a healthy diet, international nutrition committees recommend that 25–35 % of total energy intake should be from fats to prevent CVD and to regulate cholesterol homeostasis in the body. Furthermore, lowering the intakes of SFA (5–10 % of the total energy intake) and TFA (less than 1 % of the total energy intake), while replacing them with unsaturated fatty acids (MUFA and PUFA), and additionally reducing industrial trans-fats as much as possible are recommended. The literature includes mainly animal and cell culture studies, with a limited number of human studies. Extrapolation of the dose from the animals, relatively high doses, to humans needs consideration of body surface area and metabolic differences.
To combine the dietary recommendations and the novel nutrient-sensitive biomarkers related to CVD is a global focus. Thus, the current literature indicates that the type of dietary fatty acids might alter CD36 levels associated with cholesterol homeostasis. Additionally, CD36 is also a potential risk factor for hyperlipidaemia/dyslipidaemia in CD36 deficiency, relatively frequent in Asian and African populations. Nevertheless, further studies investigating cholesterol metabolism with the underlying mechanisms including CD36 and possible effects of dietary fatty acids are essential. Thus, since the CD36 receptor is suggested to be a novel nutrient-sensitive biomarker, the role of CD36 and dietary fatty acids on cholesterol metabolism might be considered in the future approving the importance in individualised medical nutrition therapy.
Acknowledgements
The present review received no specific grant from any funding agency, commercial or not-for-profit sectors.
E. U. contributed significantly to the design, screening of the articles and writing of the manuscript. R. N.-U. contributed significantly to the work’s conception, design, critical writing of the manuscript and reviewed the manuscript. Both authors read and approved the final manuscript.
The authors declare no conflicts of interest.






