Background
Governments are committed to controlling increasing healthcare expenditure, contributing to the efficiency and sustainability of healthcare systems, within a context of dwindling budgets, exacerbated by the double impact of the past economic and financial crisis beginning in 2008 and the current direct and collateral pandemic effects provoked by COVID-19. As a result, demand for evidence and justification of value, in the context of innovation and health technology assessment (HTA), are increasingly required, either to ensure efficacy and safety or, in addition, to support funding, coverage and reimbursement decisions, or price negotiations (Reference Sorenson, Drummond and Kanavos1). Although this decisional process differs among countries and even regions within countries with decentralized health systems such as Spain, Canada, or Italy, most of them use HTA, supported by clinical trial-based systematic reviews, as the main informational source (Reference Luce, Drummond, Jönsson, Neumann, Sanford-Schwartz and Siebert2;Reference Ciani and Jommi3). Additional information on health outcomes and costs under context-specific or “real-world” conditions, including outcomes data relevant for patients, is increasing but still scarce (Reference Ciani and Jommi3).
Evidence standards for HTA are threatened by incomplete and possibly misleading evidence, which is particularly present in procedures and medical devices where the evidence is usually much less extensive and robust than the evidence for drugs, given the less stringent existing regulatory requirements (Reference Maynard and Bloor4). Other domains relevant for medical device assessment, such as learning curves for capacity building, organizational issues, or environmental impact, are also commonly ignored. This situation calls for additional informational sources such as postlaunch evidence-generation (PLEG) studies, which can provide postmarketing real-world evidence (RWE), enlarging the scope of HTA in the lifecycle of technologies according to the new HTA definition (Reference Garrison, Neumann and Erickson5–Reference O'Rourke, Oortwijn and Schuller7).
The PLEG studies can be boosted by different requests and purposes. Some of them can cover academic or technology developers’ objectives. Others, however, come from a request of health authorities because of their need to rely on additional evidence to make decisions.
The healthcare system in Spain is decentralized, with a common benefit package defined at the central level with the agreement of Regional Health Authorities under the Interterritorial Council of the National Health System. The provision and financing of health care rely on the Regional Health Authorities. The Directorate General of the common portfolio of services of the National Health System (NHS) and Pharmacy (DGPSPh) of the Spanish Ministry of Health (MoH) is the national organization responsible for coverage approval of all health technologies in Spain. The Spanish Network for Health Technology Assessment and Services of the NHS (RedETS) is the organization responsible for appraising available evidence about the safety, effectiveness, cost-effectiveness, organizational aspects, and the legal, ethical, organizational, and environmental issues of all nonpharmaceutical technologies. Detailed information about the functions, composition, and activity of RedETS has been recently published by this journal (Reference Serrano-Aguilar, Asua-Batarrita, Molina-López, Espallargues, Pons-Rafols and García-Armesto8). The Spanish Agency of Medicines and Medical Devices (AEMPS) is a state agency linked to the MoH, responsible for assessing the quality, safety, and efficacy of medicines before market access of pharmacological technologies.
From 2015, the MoH launched the controlled use studies (from the Spanish: estudios de uso tutelado), a type of observational study to inform policy decision making on public funding of relevant health technologies before their inclusion in the Benefit Portfolio of the Spanish NHS (BP) (9). Later in 2015, the MoH started requesting RedETS for PLEG studies on medical devices to inform coverage decisions in the context of the NHS. These studies were officially regulated in an MoH order SSI/1356/2015 (10), which establishes the aims and main characteristics of the so-called “Estudios de Monitorización” in Spanish (i.e., Monitoring Studies [MS]). MS are prospective observational and single-arm studies of selected technologies at different stages of their postlaunch phase when evidence is limited. MS are aimed at technologies already included in the BP to assess specific conditions of use or effects in certain target population groups (Reference Varela-Lema, Ruano-Ravina, Mota, Ibargoyen-Roteta, Imaz and Gutiérrez-Ibarluzea11–13). MS' general purpose is reducing uncertainty while providing RWE mainly on the safety and effectiveness of the application of highly relevant technologies in the Spanish NHS under real practice circumstances. Economic and organizational impact examination are also included in MS. At present, MS are intended to support decision making on public funding of nonpharmaceutical technologies.
The coordination of MS is shared by the DGPSPh and RedETS. Although the DGPSPh develops all political, economic, administrative, organizational, and logistic tasks, the technical coordination and scientific exploitation are independently carried out by the designated RedETS’ agency, including the MS protocol development, the study design, data analyses, and final reporting. MS favor early technology accessibility to clinicians and patients, subject to RWE generation.
The aim of this publication is to describe the planned steps and procedures supporting MS in the context of RedETS, together with a summary of the operational experiences and some lessons learnt from the first wave of MS.
Topic Identification for MS
The DGPSPh of the MoH is responsible for the selection of topics for PLEG at the proposal of the National Commission of Provision, Insurance and Financing (CPAF) of the MoH, according to the Law SSI/1356/2015 (10) and the Royal Decree 1030/2006 (Reference Varela-Lema, Ruano-Ravina, Mota, Ibargoyen-Roteta, Imaz and Gutiérrez-Ibarluzea11). MS usually originate when specific relevant informational/research needs hindering decision making arise in relation to previous RedETS reports (Reference Rivero-Santana, Pérez-Ramos, García-Hernández, Perestelo-Pérez, Guerra-Rodríguez and Serrano-Aguilar14–Reference Imaz-Iglesia, García-Pérez, Nachtnebel, Martín-Águeda, Sánchez-Piedra and Karadayi16). The CPAF proposal must be accompanied by a clear indication of the fulfillment of the criteria to select and prioritize health technologies requiring additional evidence-generation indication, according to the specific EUnetHTA recommendations delivered by 2012 (Reference Guzina, Carbonneil and Lee-Robin17). The proposals are carefully considered given the scientific and technical complexity, time constraints and costs of the procedure, as well as the necessary strategic involvement of several healthcare centers across different Spanish autonomous regions, scientific societies, related industries, and patient associations. Each MS is assigned to a RedETS’ agency that will be responsible for conducting the technical and scientific part of the PLEG study.
Design, Protocol Structure, and Contents of MS
An MS is the specific PLEG study format devoted to gather RWE in the Spanish NHS context in the field of medical devices. It corresponds to observational single-arm studies that prospectively collect evidence about the real performance of medical devices in the Spanish NHS. All MS are applied under an investigational protocol, limiting the provision of the assessed technology and guiding its indication to a previously selected set of referral centers.
The MS protocols are the core documents describing the design, objectives, methodology, and overall organization of the study, guiding all activities of participant research groups. Each MS protocol is prepared by the designated agency and consists of two sections (9). The first section includes: (i) a health technology description covering the application procedures and organizational arrangements needed, together with the diffusion degree of the studied health technology in Spain and other EU countries, as well as comparative information with competing available alternatives. (ii) A synopsis on the target health condition and its current standards of care; the main targeted population and potential subgroups of interest; relevant contextual organizational issues related to health technology acquisition, commissioning, and operation; as well as costing, social, and ethical issues related to the technology. This part will end with the main arguments justifying the development of the MS (Reference Guzina, Carbonneil and Lee-Robin17), including the knowledge gaps and research needs to address, as well as potential worldwide-related studies currently underway. (iii) The objectives of MS. (iv) Indications of use of the technology. (v) A subsection with the conditions of use of the technology under evaluation to allow appropriate selection of participant referral centers, including all material, human, legal, and educational/training requirements for appropriate technology application.
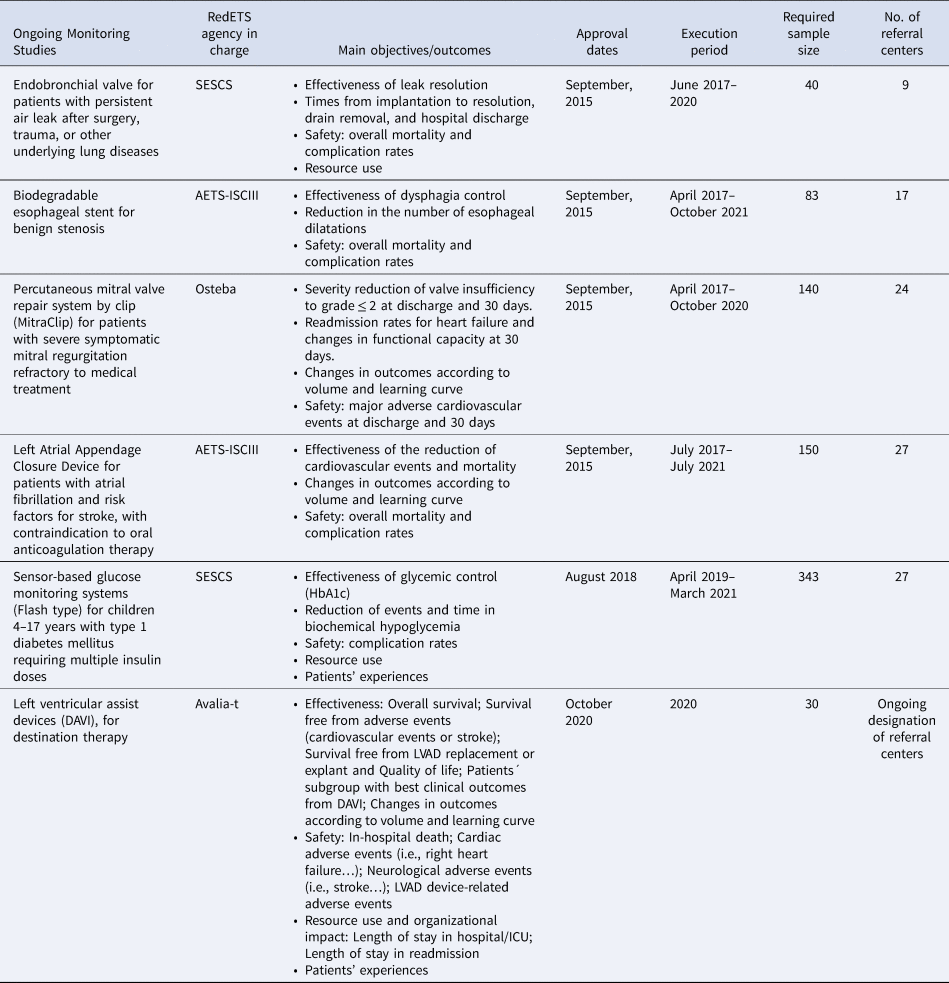
The second section covers the methodological and implementation issues of MS, including: (i) patient eligibility criteria. (ii) Minimum data set requirements, including sociodemographic and clinical information, use of healthcare resources and health outcomes accounting for favorable and unfavorable effects, with special interest for relevant predefined subgroups of patients (see Table 1 with outcome measures currently used in ongoing MS). (iii) The need for short, medium, or long-term follow-up. (iv) Sample-size estimations and expected MS duration. MS duration will vary according to sample-size requirements, condition prevalence, the number of participant referral centers, characteristics of the assessed outcomes, and estimated follow-up visits. (v) A specific informed consent form is applied and signed by all participants.
Table 1. Current MS by a commissioned RedETS agency, objectives, dates, and operational issues
Data gathering is supported by an electronic information system (SIEM) embedded into a web platform developed and managed by the DGPSPh (18). The SIEM hosts a data collection form for the initial stage, the intervention phase, and the monitoring period. Patient identification data are coded and accessible only to participant centers (referral centers), ensuring patient anonymity. Several types of automated warnings help identify relevant health events related to safety, as well as missing or improperly entered data.
Protocol development is open to participation of experts nominated by regional health authorities in the Spanish NHS and related scientific societies. Additionally, the study protocol is revised by interested patient organizations and industry representatives. All experts, patients, and professionals participating in any type of MS activity complete an individual conflict of interest declaration. Finally, the reviewed protocol is submitted for approval by the CPAF.
Due to the absence of validated quality standards to assess registries providing RWE for HTA purposes at the time of launching the first MS, RedETS relied on the general guidelines for comparable development of patient registries, proposed by the EU-funded Joint Action “Cross Border Patient Registries Initiative” (PARENT) (Reference Meglič, Doupi, Pristaš, Skalkidis, Zaletel and Orel19). RedETS soon aligned its MS criteria of quality checking to those suggested by EUnetHTA JA3 in the REQueST framework (20).
Selection of Referral Healthcare Centers
The selection of eligible healthcare centers, commonly tertiary hospitals across all Spanish autonomous communities, begins after the approval of the protocol by the CPAF (10). This is a decentralized selection process happening at the regional level under the control of every local health authority. Selected hospitals are checked and confirmed by the MoH according to the information provided by an eligibility self-assessment questionnaire, developed between RedETS and the DGPSPh. A large number of referral centers will be required when monitoring a highly needed technology, when a wider sample size is required and when assessing technologies devoted to low-prevalence clinical conditions.
Operational Procedures and Intended Use of Results
Every RedETS agency in charge of an MS has the following tasks related to its execution (10): (i) receiving and analyzing data from all participating centers. (ii) Monthly checking on SIEMs' data completeness and quality. (iii) Immediate communication to DGPSPh of all potentially technology related adverse events to consider possible changes in MS protocol or even MS discontinuation. (iv) Ensuring that data collection and transmission comply with legal requirements on protection of personal data. (v) Reviewing the informed consent forms to verify the information provided to patients. (vi) Annual deliverance of a technical report for the CPAF, updating the information on the MS evolution. Early communication of relevant recruitment delays threatening the desired sample size is highlighted, together with a realistic suggestion of expanded period when needed.
Within 3 months after completing the SIEMs' data registration, the corresponding agency prepares the preliminary final technical report by processing, analyzing, and interpreting all SIEM-contained information for external expert and industry review. The final report is submitted to DGPSPh and used for decision making by the CPAF regarding the status of the technology evaluated in the common benefit package, according to the report's conclusions and other factors such as the availability of therapeutic alternatives and its estimated organizational and economic impact. Decisions could be aimed at keeping the technology in the benefit package under the previous conditions of use, modifying these conditions of use, or even excluding the technology from public funding. Whatever the decision, its implementation requires the publication of a ministerial order, according to the Royal Decree 1030/2006. Complementary actions could also be proposed, such as keeping the technology use in selected centers or making recommendations regarding the reorganization of resources.
Once completed and approved by the CPAF, all final reports will be hosted on the RedETS Web site and shared with interested scientific and patient organizations, as well as with related industry. Subsequently, those providing scientifically relevant results will be disclosed in appropriate scientific meetings and international journals.
The Role of the Industry
Industry representatives have early access to both the preliminary MS protocol and the preliminary final report, to provide documented feedback. The DGPSPh of the MoH, together with authorities of the participant autonomous communities involved in MS, negotiates the supply and maximum price conditions of the devices under assessment with the commercializing companies, requesting them to provide a quarterly communication of the number of devices/equipment supplied to each participant center. This periodic information is sequentially used to check the progression of included cases in each participant center in SIEM. Companies are also committed to providing all new relevant information published on the monitored health technology. The RedETS MS program considers the possibility of using shared financing mechanisms with the industry (10).
First RedETS Experiences on Health Technologies under MS
The first MS of RedETS have been funded collaboratively between the MoH and the regional health authorities. Although regional health authorities support participant hospitals by reimbursing all involved direct costs, including the technology under assessment and data gathering, the MoH funds the RedETS participation to guarantee monitoring and quality assurance processes, as well as data analyses and report delivery.
Table 1 shows the main general characteristics of all MS currently monitored by RedETS. In July 2015, the MoH commissioned the first four MS (Reference Rivero-Santana, Pérez-Ramos, García-Hernández, Perestelo-Pérez, Guerra-Rodríguez and Serrano-Aguilar14–Reference Imaz-Iglesia, García-Pérez, Nachtnebel, Martín-Águeda, Sánchez-Piedra and Karadayi16). Later, two other MS were approved and commissioned; all of them related to previous HTA reports (Reference Perestelo-Pérez, Rivero-Santana, García-Lorenzo, Castilla-Rodríguez, Vallejo-Torres and García-Pérez21;Reference Puñal Riobóo and Varela Lema22).
Lessons Learnt and Implications at the National and European Levels
This article is the first of its kind to describe the real-world experiences of PLEG for medical devices, supported by national and regional health authorities in the context of the Spanish NHS. Its specific characteristics, including an officially established national procedure, supported and funded by the MoH in cooperation with regional health authorities, explain the potential interest of this MS procedure. Although the final reports of first-generation MS are currently being delivered, awaiting an impact assessment, this paper includes an analysis of difficulties to overcome to improve the selection, design, implementation, and timely execution of future MS.
As commonly happens with medical devices, all technologies under the MS program reached the market with specific and relevant scientific gaps hampering informed decisions on public coverage and reimbursement by Spanish health authorities. Even though medical devices are launched into the market with more scientific uncertainties than medicines, the use of PLEG for decision making has been more frequently explored for medicines than for other technologies. This situation is influenced by the unbalanced requirements that regulators make when uncertainties at approval are important enough to potentially have an impact on the product information and clinical use (Reference Moseley, Vamvakas, Berntgen, Cave, Kurz and Arlett23). Medical devices may be particularly affected by current efforts to promote early assessment of new short lifecycle technologies, thereby threatening fair decision making given the common scarcity, incompleteness, and quality limitations of available evidence.
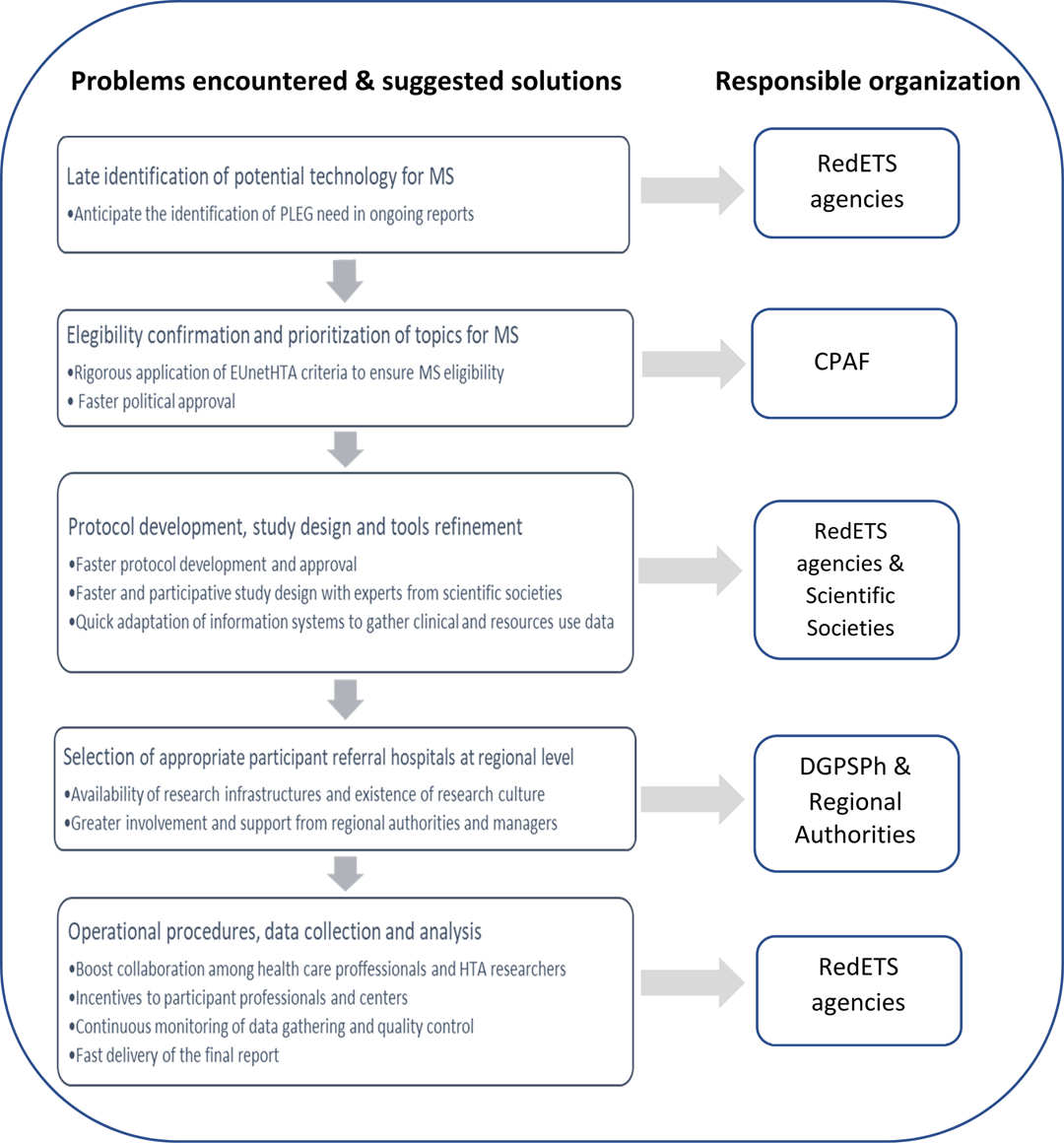
Despite the value attributed to RWE to inform national decision making in the Spanish NHS, the commitment and participation of the MoH and all regional health authorities, and the adherence of the MS to the essential quality standards for registries recently agreed by EUnetHTA (20), several relevant problems were identified by the DGPSPh, in a recent overall review of the MS processes. Although the first wave of MS required an average of 18 months from political approval to start, plus three additional years to deliver the final report, second wave MS significantly reduced, almost by half, the starting time. The main reasons explaining delays to start selected MS have to do with the process of political approval of MS proposals and their protocols, as well as with the later adaptation of the SIEM and the selection of participant referral hospitals. Besides problems in recruiting participant referral hospitals, their limited availability of research infrastructure and professional involvement also contributed to delays in the recruitment of patients, in reaching the required sample size and in the adequate and timely data collection. From this recent overall review process promoted by the MoH to improve the next generation of MS, the following needs of actions were identified: (i) to improve the selection and prioritization of technologies to include in MS; (ii) to anticipate earlier identification and inclusion of new devices similar to those under MS; (iii) to streamline all administrative tasks—shortening times, avoiding unnecessary paperwork, and setting a maximum target time of 7 months from approval to beginning of MS; (iv) to reinforce the role of regional authorities in selecting referral centers with existing research infrastructure; (v) to boost earlier and more intense collaboration among RedETS, clinical experts, patients, and industry representatives in protocol development and SIEM adaptation; and (vi) to provide incentives and intensify communication and feedback with all involved professionals to improve patient recruitment and quality and timely data collection to allow appropriate analysis and earlier delivery of the final report. Figure 1 illustrates the main problems detected, the possible solutions, and the corresponding organizations responsible for implementation.
Figure 1. Problems encountered and suggested solutions by responsible organizations. The main problems encountered and the possible actions to minimize their impact affect most of the main phases of the MS and the stakeholders involved in the selection, approval, design, and execution of MS, as well as the analysis and final reporting.
The systematic data collection (Reference Black and Payne24) of patients' features, relevant health outcomes, and resource use, under real-world scenarios subject to quality control monitoring, reduces uncertainty in national and regional coverage decision making in context-specific settings (Reference Black25;Reference Grilli and Taroni26). After technology inclusion, additional evidence regarding diffusion and patterns of clinical use in real practice could also be potentially monitored, providing valuable information on longer-term outcomes either to confirm or reverse previous decisions or to help define its appropriate implementation and use from an organizational and clinical perspective, respectively. The production of RWE, specifically for devices and surgical interventions, should consider the potentially modifying effect of the volume and the learning curve of the operator.
Grilli and Taroni (Reference Grilli and Taroni26) reported that some observational studies on emergent technologies showed that evidence collected on routine use did not always support earlier trial published outcomes, providing lower outcome values than those coming from experimental studies. Real-world settings and patients might differ from experimental studies, according to age and comorbidity distribution. Additionally, local patterns of clinical practice might diverge from experimental protocols by merging other interventions (Reference Grilli and Taroni26). Regardless of the hierarchy of evidence of observational MS, these are intended to supplement the insufficient evidence provided from available clinical trials, either when the evidence base to support decisions is immature or when there is substantial uncertainty surrounding the effectiveness, safety, or cost-effectiveness, by measuring relevant outcomes for patients under real-world conditions (Reference Claxton, Palmer, Longworth, Bojke, Griffin and McKenna27). In addition, information about uncertainties stemming from surrogate end points, long-term efficacy, effects in specific subgroups, a product's use in clinical practice (e.g., its place in the treatment algorithm; treatment duration and adherence), or changes in standard of care, could be also provided (Reference Moseley, Vamvakas, Berntgen, Cave, Kurz and Arlett23).
According to Grilli and Taroni (Reference Grilli and Taroni26), “the longer it takes for coverage decisions, the greater, broader and more organized is the pressure on policy-makers.” In fact, the time required to complete the first four MS by RedETS has been longer than expected due to initial difficulties in setting the SIEMs, recruiting participant centers and patients, and in completing all required entry and sequential data. Meanwhile, concomitant industry marketing strategies activating patient and mass media expectations were observed, putting pressure on clinicians and health policy decision makers (Reference Perestelo-Pérez, Rivero-Santana, García-Lorenzo, Castilla-Rodríguez, Vallejo-Torres and García-Pérez21).
In spite of the progress made in EUnetHTA Joint Action III to perform collaborative assessments on relative effectiveness (REA) and joint early dialogues with the industry, many limited experiences have been reported on the development of collaborative PLEG studies to complement systematic reviews with context-specific RWE for the assessment and decision support regarding medical devices. However, considering that any kind of additional data collection is resource- and time-consuming, and that the process for selecting technologies for further research used to be informal and heterogeneous among settings, the EUnetHTA Joint Action 2010–2 (Reference Guzina, Carbonneil and Lee-Robin17), and others (Reference Claxton, Palmer, Longworth, Bojke, Griffin and McKenna27), promptly provided a set of selection/prioritization criteria to guide selecting technologies for which complementary studies are of real value. Besides, at present, the EUnetHTA work package 5B is developing two pilot PLEG studies on pharmaceuticals and one pilot PLEG study on medical devices, specifically on left ventricular assist devices (DAVI) for destination therapy. Some of the difficulties faced in the case of the collaborative PLEG studies are related to sharing health data among different countries. As a result, the DAVI pilot will be focused on identifying evidence gaps and setting and gathering information on a minimum data set (28). Solving data-sharing barriers and expanding experiences of collaborative registries would be of great interest for future European HTA collaborations. Similarly, in addition to the experiences on joint REA and early dialogues, collaboration in PLEG studies for medical devices could also contribute to increased EUnetHTA productivity and impact, reducing the time needed and overall costs to deliver these types of observational reports across the EU (Reference Grilli and Taroni26).
An EUnetHTA survey examining the extent to which HTA agencies use sources of RWE such as registries (Reference Mandeville, Valentic, Ivankovic, Pristas, Long and Patrick29) showed that, particularly in Europe, some agencies use clinical and administrative databases, besides clinical registries to gather additional data for HTA. Less than half, however, currently employ criteria for quality checking these data sources, in most cases defined by each organization rather than external bodies with standardized tools. The recent development of the REQueST instrument for quality assessment of previously elsewhere developed registries is expected to ease international cooperation in the development or early adaptation of registries specifically designed for HTA purposes under the EUnetHTA framework and beyond (20).
Given the increasing growth of international and national PLEG studies, similar to the RedETS MS program to provide contextual data and support local decision making, some concern emerges regarding the possibility of a progressive effort reduction by the healthcare industry to generate sufficient and valid scientific evidence for all dimensions required for HTA and approval. This concern, together with the need to limit the financial effort required by this type of PLEG studies, should give more prominence to the activities of early scientific advice between HTA organizations and the industry representatives, to ensure that all relevant required information to inform decision making is promptly provided by the industry. The financial contribution of the industry in this type of study is, consequently justified, according to the aforementioned considerations (Reference Claxton, Palmer, Longworth, Bojke, Griffin and McKenna27). Despite the fact that the RedETS MS program envisaged the potential funding contribution by the involved industry (10), no experiences of shared financial responsibilities have been currently developed by the MoH in the specific field of PLEG studies for medical devices. The main reasons explaining this initial decision lie in the convenience of developing previous experiences with simplified implementation models, in which the most relevant activities in each stage of the MS could be tested, characterized, and established. In addition, it has been possible to estimate the costs of MS, before adding administrative and financial complexities by involving the industry.
Finally, consideration is required on how the new Medical Device Regulation (MDR) at the European Union level will affect the availability and quality of data at the time of launching new health technologies. PLEG studies should have been aligned with this recently adopted MDR, but they were immediately postponed until May 2021 due to the current challenges related to the pandemic crisis (30). The new MDR was developed by focusing on patient health and safety as core principles, requiring companies to provide data on safety under real practice conditions. On such a basis, there will be a need to discuss with regional systems, professionals, patients, and companies how to adapt PLEG studies to these new requirements to better address the information needs to better support decision making on the approval and provision of new medical devices and in vitro diagnostics.
As a brief conclusion, the RedETS MS program is intended to complement and enhance the conventional HTA process to support stakeholder decision making in Spain, including health authorities, clinicians, and patients. The MS program is focused on specific uncertainties about the real effect, safety, costs, and routine use of new and insufficiently assessed relevant medical devices carefully selected to ensure the value of the additional research needed, by means of controlled, participative, and transparent procedures. However, despite a clear political commitment and economic support from national and regional health authorities, several difficulties were identified during the development and implementation of the first wave of MS, delaying its execution and final reporting, challenging their impact on political decisions. Resolution of these difficulties at the national level and a greater collaborative impulse at the European level, given the availability of an appropriate methodological framework already provided by EUnetHTA, might provide a faster and more efficient comparative RWE of improved quality and reliability.





 Open access
Open access
Background
Governments are committed to controlling increasing healthcare expenditure, contributing to the efficiency and sustainability of healthcare systems, within a context of dwindling budgets, exacerbated by the double impact of the past economic and financial crisis beginning in 2008 and the current direct and collateral pandemic effects provoked by COVID-19. As a result, demand for evidence and justification of value, in the context of innovation and health technology assessment (HTA), are increasingly required, either to ensure efficacy and safety or, in addition, to support funding, coverage and reimbursement decisions, or price negotiations (Reference Sorenson, Drummond and Kanavos1). Although this decisional process differs among countries and even regions within countries with decentralized health systems such as Spain, Canada, or Italy, most of them use HTA, supported by clinical trial-based systematic reviews, as the main informational source (Reference Luce, Drummond, Jönsson, Neumann, Sanford-Schwartz and Siebert2;Reference Ciani and Jommi3). Additional information on health outcomes and costs under context-specific or “real-world” conditions, including outcomes data relevant for patients, is increasing but still scarce (Reference Ciani and Jommi3).
Evidence standards for HTA are threatened by incomplete and possibly misleading evidence, which is particularly present in procedures and medical devices where the evidence is usually much less extensive and robust than the evidence for drugs, given the less stringent existing regulatory requirements (Reference Maynard and Bloor4). Other domains relevant for medical device assessment, such as learning curves for capacity building, organizational issues, or environmental impact, are also commonly ignored. This situation calls for additional informational sources such as postlaunch evidence-generation (PLEG) studies, which can provide postmarketing real-world evidence (RWE), enlarging the scope of HTA in the lifecycle of technologies according to the new HTA definition (Reference Garrison, Neumann and Erickson5–Reference O'Rourke, Oortwijn and Schuller7).
The PLEG studies can be boosted by different requests and purposes. Some of them can cover academic or technology developers’ objectives. Others, however, come from a request of health authorities because of their need to rely on additional evidence to make decisions.
The healthcare system in Spain is decentralized, with a common benefit package defined at the central level with the agreement of Regional Health Authorities under the Interterritorial Council of the National Health System. The provision and financing of health care rely on the Regional Health Authorities. The Directorate General of the common portfolio of services of the National Health System (NHS) and Pharmacy (DGPSPh) of the Spanish Ministry of Health (MoH) is the national organization responsible for coverage approval of all health technologies in Spain. The Spanish Network for Health Technology Assessment and Services of the NHS (RedETS) is the organization responsible for appraising available evidence about the safety, effectiveness, cost-effectiveness, organizational aspects, and the legal, ethical, organizational, and environmental issues of all nonpharmaceutical technologies. Detailed information about the functions, composition, and activity of RedETS has been recently published by this journal (Reference Serrano-Aguilar, Asua-Batarrita, Molina-López, Espallargues, Pons-Rafols and García-Armesto8). The Spanish Agency of Medicines and Medical Devices (AEMPS) is a state agency linked to the MoH, responsible for assessing the quality, safety, and efficacy of medicines before market access of pharmacological technologies.
From 2015, the MoH launched the controlled use studies (from the Spanish: estudios de uso tutelado), a type of observational study to inform policy decision making on public funding of relevant health technologies before their inclusion in the Benefit Portfolio of the Spanish NHS (BP) (9). Later in 2015, the MoH started requesting RedETS for PLEG studies on medical devices to inform coverage decisions in the context of the NHS. These studies were officially regulated in an MoH order SSI/1356/2015 (10), which establishes the aims and main characteristics of the so-called “Estudios de Monitorización” in Spanish (i.e., Monitoring Studies [MS]). MS are prospective observational and single-arm studies of selected technologies at different stages of their postlaunch phase when evidence is limited. MS are aimed at technologies already included in the BP to assess specific conditions of use or effects in certain target population groups (Reference Varela-Lema, Ruano-Ravina, Mota, Ibargoyen-Roteta, Imaz and Gutiérrez-Ibarluzea11–13). MS' general purpose is reducing uncertainty while providing RWE mainly on the safety and effectiveness of the application of highly relevant technologies in the Spanish NHS under real practice circumstances. Economic and organizational impact examination are also included in MS. At present, MS are intended to support decision making on public funding of nonpharmaceutical technologies.
The coordination of MS is shared by the DGPSPh and RedETS. Although the DGPSPh develops all political, economic, administrative, organizational, and logistic tasks, the technical coordination and scientific exploitation are independently carried out by the designated RedETS’ agency, including the MS protocol development, the study design, data analyses, and final reporting. MS favor early technology accessibility to clinicians and patients, subject to RWE generation.
The aim of this publication is to describe the planned steps and procedures supporting MS in the context of RedETS, together with a summary of the operational experiences and some lessons learnt from the first wave of MS.
Topic Identification for MS
The DGPSPh of the MoH is responsible for the selection of topics for PLEG at the proposal of the National Commission of Provision, Insurance and Financing (CPAF) of the MoH, according to the Law SSI/1356/2015 (10) and the Royal Decree 1030/2006 (Reference Varela-Lema, Ruano-Ravina, Mota, Ibargoyen-Roteta, Imaz and Gutiérrez-Ibarluzea11). MS usually originate when specific relevant informational/research needs hindering decision making arise in relation to previous RedETS reports (Reference Rivero-Santana, Pérez-Ramos, García-Hernández, Perestelo-Pérez, Guerra-Rodríguez and Serrano-Aguilar14–Reference Imaz-Iglesia, García-Pérez, Nachtnebel, Martín-Águeda, Sánchez-Piedra and Karadayi16). The CPAF proposal must be accompanied by a clear indication of the fulfillment of the criteria to select and prioritize health technologies requiring additional evidence-generation indication, according to the specific EUnetHTA recommendations delivered by 2012 (Reference Guzina, Carbonneil and Lee-Robin17). The proposals are carefully considered given the scientific and technical complexity, time constraints and costs of the procedure, as well as the necessary strategic involvement of several healthcare centers across different Spanish autonomous regions, scientific societies, related industries, and patient associations. Each MS is assigned to a RedETS’ agency that will be responsible for conducting the technical and scientific part of the PLEG study.
Design, Protocol Structure, and Contents of MS
An MS is the specific PLEG study format devoted to gather RWE in the Spanish NHS context in the field of medical devices. It corresponds to observational single-arm studies that prospectively collect evidence about the real performance of medical devices in the Spanish NHS. All MS are applied under an investigational protocol, limiting the provision of the assessed technology and guiding its indication to a previously selected set of referral centers.
The MS protocols are the core documents describing the design, objectives, methodology, and overall organization of the study, guiding all activities of participant research groups. Each MS protocol is prepared by the designated agency and consists of two sections (9). The first section includes: (i) a health technology description covering the application procedures and organizational arrangements needed, together with the diffusion degree of the studied health technology in Spain and other EU countries, as well as comparative information with competing available alternatives. (ii) A synopsis on the target health condition and its current standards of care; the main targeted population and potential subgroups of interest; relevant contextual organizational issues related to health technology acquisition, commissioning, and operation; as well as costing, social, and ethical issues related to the technology. This part will end with the main arguments justifying the development of the MS (Reference Guzina, Carbonneil and Lee-Robin17), including the knowledge gaps and research needs to address, as well as potential worldwide-related studies currently underway. (iii) The objectives of MS. (iv) Indications of use of the technology. (v) A subsection with the conditions of use of the technology under evaluation to allow appropriate selection of participant referral centers, including all material, human, legal, and educational/training requirements for appropriate technology application.
The second section covers the methodological and implementation issues of MS, including: (i) patient eligibility criteria. (ii) Minimum data set requirements, including sociodemographic and clinical information, use of healthcare resources and health outcomes accounting for favorable and unfavorable effects, with special interest for relevant predefined subgroups of patients (see Table 1 with outcome measures currently used in ongoing MS). (iii) The need for short, medium, or long-term follow-up. (iv) Sample-size estimations and expected MS duration. MS duration will vary according to sample-size requirements, condition prevalence, the number of participant referral centers, characteristics of the assessed outcomes, and estimated follow-up visits. (v) A specific informed consent form is applied and signed by all participants.
Table 1. Current MS by a commissioned RedETS agency, objectives, dates, and operational issues
SESCS, Servicio de Evaluación del Servicio Canario de la Salud; AETS-ISCIII, Agencia de Evaluación de Tecnologías Sanitarias del Instituto de Salud Carlos III; Osteba, Servicio de Evaluación de Tecnologías Sanitarias. Departamento de Salud del País Vasco; Avalia-t, Unidade de Asesoramento Científico-técnico, avalia-t. Axencia de Coñecemento en Saúde (ACIS).
Data gathering is supported by an electronic information system (SIEM) embedded into a web platform developed and managed by the DGPSPh (18). The SIEM hosts a data collection form for the initial stage, the intervention phase, and the monitoring period. Patient identification data are coded and accessible only to participant centers (referral centers), ensuring patient anonymity. Several types of automated warnings help identify relevant health events related to safety, as well as missing or improperly entered data.
Protocol development is open to participation of experts nominated by regional health authorities in the Spanish NHS and related scientific societies. Additionally, the study protocol is revised by interested patient organizations and industry representatives. All experts, patients, and professionals participating in any type of MS activity complete an individual conflict of interest declaration. Finally, the reviewed protocol is submitted for approval by the CPAF.
Due to the absence of validated quality standards to assess registries providing RWE for HTA purposes at the time of launching the first MS, RedETS relied on the general guidelines for comparable development of patient registries, proposed by the EU-funded Joint Action “Cross Border Patient Registries Initiative” (PARENT) (Reference Meglič, Doupi, Pristaš, Skalkidis, Zaletel and Orel19). RedETS soon aligned its MS criteria of quality checking to those suggested by EUnetHTA JA3 in the REQueST framework (20).
Selection of Referral Healthcare Centers
The selection of eligible healthcare centers, commonly tertiary hospitals across all Spanish autonomous communities, begins after the approval of the protocol by the CPAF (10). This is a decentralized selection process happening at the regional level under the control of every local health authority. Selected hospitals are checked and confirmed by the MoH according to the information provided by an eligibility self-assessment questionnaire, developed between RedETS and the DGPSPh. A large number of referral centers will be required when monitoring a highly needed technology, when a wider sample size is required and when assessing technologies devoted to low-prevalence clinical conditions.
Operational Procedures and Intended Use of Results
Every RedETS agency in charge of an MS has the following tasks related to its execution (10): (i) receiving and analyzing data from all participating centers. (ii) Monthly checking on SIEMs' data completeness and quality. (iii) Immediate communication to DGPSPh of all potentially technology related adverse events to consider possible changes in MS protocol or even MS discontinuation. (iv) Ensuring that data collection and transmission comply with legal requirements on protection of personal data. (v) Reviewing the informed consent forms to verify the information provided to patients. (vi) Annual deliverance of a technical report for the CPAF, updating the information on the MS evolution. Early communication of relevant recruitment delays threatening the desired sample size is highlighted, together with a realistic suggestion of expanded period when needed.
Within 3 months after completing the SIEMs' data registration, the corresponding agency prepares the preliminary final technical report by processing, analyzing, and interpreting all SIEM-contained information for external expert and industry review. The final report is submitted to DGPSPh and used for decision making by the CPAF regarding the status of the technology evaluated in the common benefit package, according to the report's conclusions and other factors such as the availability of therapeutic alternatives and its estimated organizational and economic impact. Decisions could be aimed at keeping the technology in the benefit package under the previous conditions of use, modifying these conditions of use, or even excluding the technology from public funding. Whatever the decision, its implementation requires the publication of a ministerial order, according to the Royal Decree 1030/2006. Complementary actions could also be proposed, such as keeping the technology use in selected centers or making recommendations regarding the reorganization of resources.
Once completed and approved by the CPAF, all final reports will be hosted on the RedETS Web site and shared with interested scientific and patient organizations, as well as with related industry. Subsequently, those providing scientifically relevant results will be disclosed in appropriate scientific meetings and international journals.
The Role of the Industry
Industry representatives have early access to both the preliminary MS protocol and the preliminary final report, to provide documented feedback. The DGPSPh of the MoH, together with authorities of the participant autonomous communities involved in MS, negotiates the supply and maximum price conditions of the devices under assessment with the commercializing companies, requesting them to provide a quarterly communication of the number of devices/equipment supplied to each participant center. This periodic information is sequentially used to check the progression of included cases in each participant center in SIEM. Companies are also committed to providing all new relevant information published on the monitored health technology. The RedETS MS program considers the possibility of using shared financing mechanisms with the industry (10).
First RedETS Experiences on Health Technologies under MS
The first MS of RedETS have been funded collaboratively between the MoH and the regional health authorities. Although regional health authorities support participant hospitals by reimbursing all involved direct costs, including the technology under assessment and data gathering, the MoH funds the RedETS participation to guarantee monitoring and quality assurance processes, as well as data analyses and report delivery.
Table 1 shows the main general characteristics of all MS currently monitored by RedETS. In July 2015, the MoH commissioned the first four MS (Reference Rivero-Santana, Pérez-Ramos, García-Hernández, Perestelo-Pérez, Guerra-Rodríguez and Serrano-Aguilar14–Reference Imaz-Iglesia, García-Pérez, Nachtnebel, Martín-Águeda, Sánchez-Piedra and Karadayi16). Later, two other MS were approved and commissioned; all of them related to previous HTA reports (Reference Perestelo-Pérez, Rivero-Santana, García-Lorenzo, Castilla-Rodríguez, Vallejo-Torres and García-Pérez21;Reference Puñal Riobóo and Varela Lema22).
Lessons Learnt and Implications at the National and European Levels
This article is the first of its kind to describe the real-world experiences of PLEG for medical devices, supported by national and regional health authorities in the context of the Spanish NHS. Its specific characteristics, including an officially established national procedure, supported and funded by the MoH in cooperation with regional health authorities, explain the potential interest of this MS procedure. Although the final reports of first-generation MS are currently being delivered, awaiting an impact assessment, this paper includes an analysis of difficulties to overcome to improve the selection, design, implementation, and timely execution of future MS.
As commonly happens with medical devices, all technologies under the MS program reached the market with specific and relevant scientific gaps hampering informed decisions on public coverage and reimbursement by Spanish health authorities. Even though medical devices are launched into the market with more scientific uncertainties than medicines, the use of PLEG for decision making has been more frequently explored for medicines than for other technologies. This situation is influenced by the unbalanced requirements that regulators make when uncertainties at approval are important enough to potentially have an impact on the product information and clinical use (Reference Moseley, Vamvakas, Berntgen, Cave, Kurz and Arlett23). Medical devices may be particularly affected by current efforts to promote early assessment of new short lifecycle technologies, thereby threatening fair decision making given the common scarcity, incompleteness, and quality limitations of available evidence.
Despite the value attributed to RWE to inform national decision making in the Spanish NHS, the commitment and participation of the MoH and all regional health authorities, and the adherence of the MS to the essential quality standards for registries recently agreed by EUnetHTA (20), several relevant problems were identified by the DGPSPh, in a recent overall review of the MS processes. Although the first wave of MS required an average of 18 months from political approval to start, plus three additional years to deliver the final report, second wave MS significantly reduced, almost by half, the starting time. The main reasons explaining delays to start selected MS have to do with the process of political approval of MS proposals and their protocols, as well as with the later adaptation of the SIEM and the selection of participant referral hospitals. Besides problems in recruiting participant referral hospitals, their limited availability of research infrastructure and professional involvement also contributed to delays in the recruitment of patients, in reaching the required sample size and in the adequate and timely data collection. From this recent overall review process promoted by the MoH to improve the next generation of MS, the following needs of actions were identified: (i) to improve the selection and prioritization of technologies to include in MS; (ii) to anticipate earlier identification and inclusion of new devices similar to those under MS; (iii) to streamline all administrative tasks—shortening times, avoiding unnecessary paperwork, and setting a maximum target time of 7 months from approval to beginning of MS; (iv) to reinforce the role of regional authorities in selecting referral centers with existing research infrastructure; (v) to boost earlier and more intense collaboration among RedETS, clinical experts, patients, and industry representatives in protocol development and SIEM adaptation; and (vi) to provide incentives and intensify communication and feedback with all involved professionals to improve patient recruitment and quality and timely data collection to allow appropriate analysis and earlier delivery of the final report. Figure 1 illustrates the main problems detected, the possible solutions, and the corresponding organizations responsible for implementation.
Figure 1. Problems encountered and suggested solutions by responsible organizations. The main problems encountered and the possible actions to minimize their impact affect most of the main phases of the MS and the stakeholders involved in the selection, approval, design, and execution of MS, as well as the analysis and final reporting.
The systematic data collection (Reference Black and Payne24) of patients' features, relevant health outcomes, and resource use, under real-world scenarios subject to quality control monitoring, reduces uncertainty in national and regional coverage decision making in context-specific settings (Reference Black25;Reference Grilli and Taroni26). After technology inclusion, additional evidence regarding diffusion and patterns of clinical use in real practice could also be potentially monitored, providing valuable information on longer-term outcomes either to confirm or reverse previous decisions or to help define its appropriate implementation and use from an organizational and clinical perspective, respectively. The production of RWE, specifically for devices and surgical interventions, should consider the potentially modifying effect of the volume and the learning curve of the operator.
Grilli and Taroni (Reference Grilli and Taroni26) reported that some observational studies on emergent technologies showed that evidence collected on routine use did not always support earlier trial published outcomes, providing lower outcome values than those coming from experimental studies. Real-world settings and patients might differ from experimental studies, according to age and comorbidity distribution. Additionally, local patterns of clinical practice might diverge from experimental protocols by merging other interventions (Reference Grilli and Taroni26). Regardless of the hierarchy of evidence of observational MS, these are intended to supplement the insufficient evidence provided from available clinical trials, either when the evidence base to support decisions is immature or when there is substantial uncertainty surrounding the effectiveness, safety, or cost-effectiveness, by measuring relevant outcomes for patients under real-world conditions (Reference Claxton, Palmer, Longworth, Bojke, Griffin and McKenna27). In addition, information about uncertainties stemming from surrogate end points, long-term efficacy, effects in specific subgroups, a product's use in clinical practice (e.g., its place in the treatment algorithm; treatment duration and adherence), or changes in standard of care, could be also provided (Reference Moseley, Vamvakas, Berntgen, Cave, Kurz and Arlett23).
According to Grilli and Taroni (Reference Grilli and Taroni26), “the longer it takes for coverage decisions, the greater, broader and more organized is the pressure on policy-makers.” In fact, the time required to complete the first four MS by RedETS has been longer than expected due to initial difficulties in setting the SIEMs, recruiting participant centers and patients, and in completing all required entry and sequential data. Meanwhile, concomitant industry marketing strategies activating patient and mass media expectations were observed, putting pressure on clinicians and health policy decision makers (Reference Perestelo-Pérez, Rivero-Santana, García-Lorenzo, Castilla-Rodríguez, Vallejo-Torres and García-Pérez21).
In spite of the progress made in EUnetHTA Joint Action III to perform collaborative assessments on relative effectiveness (REA) and joint early dialogues with the industry, many limited experiences have been reported on the development of collaborative PLEG studies to complement systematic reviews with context-specific RWE for the assessment and decision support regarding medical devices. However, considering that any kind of additional data collection is resource- and time-consuming, and that the process for selecting technologies for further research used to be informal and heterogeneous among settings, the EUnetHTA Joint Action 2010–2 (Reference Guzina, Carbonneil and Lee-Robin17), and others (Reference Claxton, Palmer, Longworth, Bojke, Griffin and McKenna27), promptly provided a set of selection/prioritization criteria to guide selecting technologies for which complementary studies are of real value. Besides, at present, the EUnetHTA work package 5B is developing two pilot PLEG studies on pharmaceuticals and one pilot PLEG study on medical devices, specifically on left ventricular assist devices (DAVI) for destination therapy. Some of the difficulties faced in the case of the collaborative PLEG studies are related to sharing health data among different countries. As a result, the DAVI pilot will be focused on identifying evidence gaps and setting and gathering information on a minimum data set (28). Solving data-sharing barriers and expanding experiences of collaborative registries would be of great interest for future European HTA collaborations. Similarly, in addition to the experiences on joint REA and early dialogues, collaboration in PLEG studies for medical devices could also contribute to increased EUnetHTA productivity and impact, reducing the time needed and overall costs to deliver these types of observational reports across the EU (Reference Grilli and Taroni26).
An EUnetHTA survey examining the extent to which HTA agencies use sources of RWE such as registries (Reference Mandeville, Valentic, Ivankovic, Pristas, Long and Patrick29) showed that, particularly in Europe, some agencies use clinical and administrative databases, besides clinical registries to gather additional data for HTA. Less than half, however, currently employ criteria for quality checking these data sources, in most cases defined by each organization rather than external bodies with standardized tools. The recent development of the REQueST instrument for quality assessment of previously elsewhere developed registries is expected to ease international cooperation in the development or early adaptation of registries specifically designed for HTA purposes under the EUnetHTA framework and beyond (20).
Given the increasing growth of international and national PLEG studies, similar to the RedETS MS program to provide contextual data and support local decision making, some concern emerges regarding the possibility of a progressive effort reduction by the healthcare industry to generate sufficient and valid scientific evidence for all dimensions required for HTA and approval. This concern, together with the need to limit the financial effort required by this type of PLEG studies, should give more prominence to the activities of early scientific advice between HTA organizations and the industry representatives, to ensure that all relevant required information to inform decision making is promptly provided by the industry. The financial contribution of the industry in this type of study is, consequently justified, according to the aforementioned considerations (Reference Claxton, Palmer, Longworth, Bojke, Griffin and McKenna27). Despite the fact that the RedETS MS program envisaged the potential funding contribution by the involved industry (10), no experiences of shared financial responsibilities have been currently developed by the MoH in the specific field of PLEG studies for medical devices. The main reasons explaining this initial decision lie in the convenience of developing previous experiences with simplified implementation models, in which the most relevant activities in each stage of the MS could be tested, characterized, and established. In addition, it has been possible to estimate the costs of MS, before adding administrative and financial complexities by involving the industry.
Finally, consideration is required on how the new Medical Device Regulation (MDR) at the European Union level will affect the availability and quality of data at the time of launching new health technologies. PLEG studies should have been aligned with this recently adopted MDR, but they were immediately postponed until May 2021 due to the current challenges related to the pandemic crisis (30). The new MDR was developed by focusing on patient health and safety as core principles, requiring companies to provide data on safety under real practice conditions. On such a basis, there will be a need to discuss with regional systems, professionals, patients, and companies how to adapt PLEG studies to these new requirements to better address the information needs to better support decision making on the approval and provision of new medical devices and in vitro diagnostics.
As a brief conclusion, the RedETS MS program is intended to complement and enhance the conventional HTA process to support stakeholder decision making in Spain, including health authorities, clinicians, and patients. The MS program is focused on specific uncertainties about the real effect, safety, costs, and routine use of new and insufficiently assessed relevant medical devices carefully selected to ensure the value of the additional research needed, by means of controlled, participative, and transparent procedures. However, despite a clear political commitment and economic support from national and regional health authorities, several difficulties were identified during the development and implementation of the first wave of MS, delaying its execution and final reporting, challenging their impact on political decisions. Resolution of these difficulties at the national level and a greater collaborative impulse at the European level, given the availability of an appropriate methodological framework already provided by EUnetHTA, might provide a faster and more efficient comparative RWE of improved quality and reliability.
Acknowledgments
The authors wish to thank Patrick Dennis for proofreading the report.
Funding
No specific funding has been required to develop this report. The financial support for the activities of RedETS is provided by the Spanish Ministry of Health.
Conflicts of Interest
The authors declare that they have no conflicts of interest.