


Colorectal cancer (CRC) has emerged as one of the most prevalent types of cancer worldwide and is the fourth most common cause of cancer mortality(Reference Ferlay, Shin and Bray1). CRC incidence varies considerably across geographical regions. It is the highest (approximately 20–45 per 100 000) among affluent societies such as Australia/New Zealand, Europe, the USA and the UK, and the lowest among African and Asian countries (approximately 5–20 per 100 000)(Reference Ferlay, Shin and Bray1). Japan currently records the highest incidence of CRC and this appears likely to persist and continue over time(Reference Center, Jemal and Smith2, Reference Parkin, Bray and Ferlay3). In countries such as Korea, Singapore and Eastern Europe, the incidence of disease is approaching that of high-risk countries with a longer history of affluence(Reference Center, Jemal and Smith2, Reference Haggar and Boushey4, Reference Wong and Eu5). This increase in the risk of CRC has been attributed to industrialisation and accompanying environmental influences associated with a transition from a low- to high-income economy(Reference Center, Jemal and Smith2). Epidemiological studies have drawn strong correlations between CRC incidence and modifiable lifestyle factors such as body weight, diet, physical activity, smoking and alcohol consumption(Reference Haggar and Boushey4). These associations indicate the strong possibility of CRC prevention and it is believed that 30–60 % of cases can be prevented with appropriate nutrition and diet(6, Reference Platz, Willett and Colditz7).
Dietary fibre is one of the most promising candidates for a protective role in CRC, with strong support from epidemiological and experimental animal studies. However, there is a degree of ambiguity in the population data, with some studies showing no significant effect(Reference Bingham, Day and Luben8, Reference Park, Hunter and Spiegelman9). This may reflect the food sources of fibre. Recently, the European Prospective Investigation into Cancer and Nutrition reported that although total dietary fibre was associated with a 30 % reduction in CRC risk, no one food source offered more protection than another(Reference Bingham, Day and Luben8). While fibre is derived largely from plant foods, it must be recognised that any protective effects of particular fibre-containing food subgroups (e.g. fruits and vegetables) can also be contributed by other nutrients present such as β-carotene, lycopene and polyphenols(Reference McCullough, Robertson and Chao10). Dietary fibre consists largely of plant polysaccharides that resist human small-intestinal enzymes and some of the protection against colorectal tumorigenesis may reflect this bulking action. Greater stool mass is expected to lower the exposure of colonocytes to carcinogens and mutagens through physical dilution and also through reduction in transit time. There is also a strong case for protection through the interactions between the large-bowel microbiome and fibre polysaccharides, which are emerging as a critical factor in the promotion of optimal colonic function(Reference Brouns, Kettlitz and Arrigoni11, Reference Topping and Clifton12). As in obligate herbivores, so too in humans, there is substantial microbial fermentation of fibre with SCFA, primarily acetate, propionate and butyrate, as significant end products(Reference Topping and Clifton12, Reference Bugaut13). The rate of fermentation of fibre varies according to type and food source; e.g. fibre derived from grains is fermented much more slowly and less completely than that from fruits and vegetables(Reference Freudenheim, Graham and Horvath14, Reference Stephen and Cummings15). Cellulose is a major constituent of plant cell walls in both cereals and fruits and vegetables, but there are major differences between the two groups. Soluble fibre polysaccharides are generally higher in fruits and vegetables, reflecting their higher content of uronic acids. In contrast, cereal grains contain more arabinoxylans, mixed-linkage glucans and oligosaccharides(Reference Marlett16, Reference Topping17). There is also a major difference between the various fibre polysaccharides (e.g. NSP and resistant starch (RS)) in the profile of SCFA which are produced. In the diets of industrialised countries at high risk of CRC (e.g. Australia), fibre intake is largely as cereal NSP with relatively little RS. In contrast, it appears that in traditional agrarian societies at low risk, NSP intakes are comparatively low but RS intakes are high. This is likely to be a significant factor in disease as RS fermentation favours butyrate production over that of NSP and large-bowel butyrate levels are higher when foods common to such populations are consumed(Reference Ahmed, Segal and Hassan18). Importantly, butyrate is the primary energy source for colonocytes where its oxidation contributes to at least 60 % of the cell's energy requirements(Reference Cummings19) and it is the SCFA most associated with protection against colorectal carcinogenesis(Reference Perrin, Pierre and Patry20).
This review focuses on how butyrate, a SCFA produced in the colon by the fermentation of dietary fibre, potentially prevents colorectal oncogenesis. Here, we review the experimental data derived from in vitro and animal model systems describing the mechanisms, in addition to histone deacetylase (HDAC) inhibition, influencing butyrate's anti-tumorigenic actions. Taken together, these data provide compelling evidence to support human intervention studies to determine the true potential of RS in lowering the risk of colorectal oncogenesis in the wider population.
Anti-tumorigenic properties of butyrate
SCFA play a significant role in maintaining the normal physiological functions of the colonic mucosa(Reference Topping and Clifton12) (Fig. 1). Although acetate is the most abundant colonic SCFA, butyrate has been studied the most due to its potent anti-tumorigenic properties. Butyrate inhibits proliferation and induces differentiation and apoptosis of CRC cells in vitro at concentrations similar to those found in the large bowel in vivo. Increased butyrate supply reduces the incidence of carcinogen-induced colon tumours in rodent models, partly through induction of apoptosis(Reference Clarke, Topping and Bird21, Reference Le Leu, Hu and Brown22). It also opposes diet-induced colonocyte DNA damage in animals, supporting its potential to promote genetic stability(Reference Toden, Bird and Topping23). Animal experiments have shown that consumption of red meat induces the formation of N-nitroso-compounds and DNA adducts, in particular the O6-methyl-adduct of 2′-deoxyguanosine(Reference Lewin, Bailey and Bandaletova24). Moreover, it has been shown that tissues display different abilities in the removal of these adducts(Reference Tan, Gerber and Cosgrove25). Failure to remove these adducts via either intrinsic DNA repair mechanisms or apoptosis results in the elevation of mutation rates. Where mutations occur in key oncogenes (e.g. Kirsten rat sarcoma viral oncogene homolog; KRAS) or tumour suppressor genes (e.g. p53), the risk for CRC development can rise dramatically(Reference Kampman, Voskuil and van Kraats26, Reference Slattery, Curtin and Anderson27).
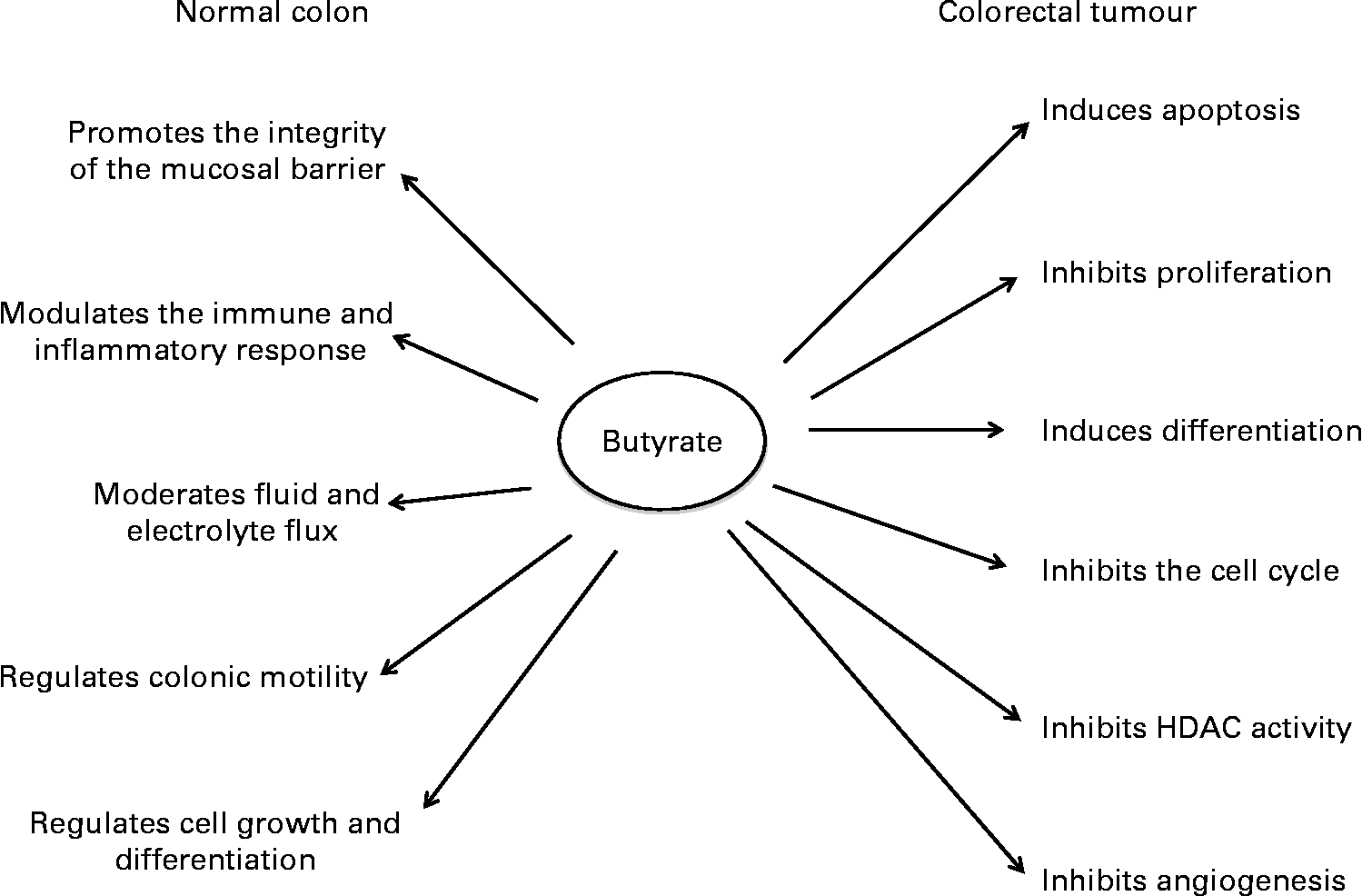
Fig. 1 The effects of butyrate in the normal colon and in colorectal tumour cells. HDAC, histone deacetylase.
The effects of butyrate are highly selective for cancer cells and its ability to modulate numerous cellular processes has contributed to the difficulty in identifying the precise mechanisms underlying each of its anti-tumorigenic properties, in particular the ability to induce apoptosis. Some of the key molecules mediating butyrate's action are summarised in Table 1. Many proteomic and transcriptomic studies have been conducted to understand the signalling pathways involved in butyrate-induced apoptosis(Reference Daly and Shirazi-Beechey28–Reference Tan, Seow and Liang33) and also to understand the mechanisms involved in the development of butyrate resistance; i.e. how a sub-population of cancer cells circumvents apoptosis in a butyrate-rich environment to form tumours(Reference Fung, Brierley and Henderson29, Reference Fung, Lewanowitsch and Henderson30, Reference Olmo, Turnay and Perez-Ramos34). Studies have shown that CRC cells that are glycolytic and have adapted to metabolise butyrate in a high-butyrate environment; i.e. butyrate-resistant cells have a growth advantage and potentially form more aggressive cancers(Reference Lopez de Silanes, Olmo and Turnay35, Reference Serpa, Caiado and Carvalho36). Although the underlying mechanisms and signalling pathways leading to these observed changes remain elusive, the recent discovery of receptors with affinity for butyrate and other SCFA strengthens the possibility that butyrate-induced apoptosis is mediated by mechanisms in addition to HDAC inhibition.
Table 1 Summary of the genes and proteins involved in the anti-tumorigenic effects of butyrate
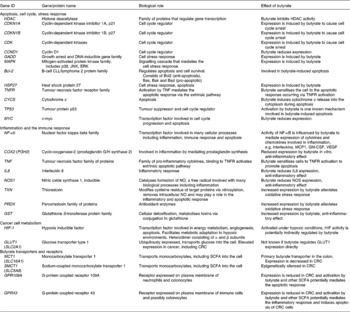
JNK, c-Jun N-terminal kinase; ERK, extracellular signal-regulated kinase; MCP1, monocyte chemoattractant protein 1; GM-CSF, granulocyte-macrophage colony stimulating factor; VEGF, vascular endothelial growth factor; CRC, colorectal cancer.
Mechanisms of butyrate-induced apoptosis in colorectal cancer cells
Studies focused on the mechanisms involved in butyrate-induced apoptosis have consistently demonstrated a rapid release of cytochrome c into the cytosol and activation of the caspase cascade as key features. In vitro studies have demonstrated that butyrate (>0·5 mmol/l) induces apoptosis in CRC cell lines(Reference Kim, Park and Lee37). Butyrate is reported to activate the intrinsic pathway of apoptosis, to sensitise cancer cells to apoptosis mediated by the extrinsic pathway and, more recently, butyrate has been shown to induce autophagic cell death(Reference Pajak, Gajkowska and Orzechowski38–Reference Wang, Luo and Xia41). These pathways appear to be activated in parallel to amplify the apoptotic response. Dysregulated expressions, resulting from butyrate treatment of pro- and anti-apoptotic proteins such as those belonging to the Bcl-2 protein family(Reference Avivi-Green, Polak-Charcon and Madar42, Reference Ruemmele, Dionne and Qureshi43) or the TNF receptor superfamily have also been reported(Reference Kim, Park and Lee37, Reference Pajak, Gajkowska and Orzechowski38).
Although activation of these cascades by butyrate has been demonstrated consistently, the triggers responsible for their initiation remain elusive. Butyrate (at 2–4 mmol/l) elicits a cell stress response in vitro characterised by the activation of genes such as those belonging to the growth arrest and DNA damage-inducible (GADD) family and activation of the mitogen-activated protein kinase (MAPK) signalling pathway(Reference Scott, Longpre and Loo44–Reference Zhang, Zhou and Bao46). A report by Scott et al. (Reference Scott, Longpre and Loo44) correlated MAPK activation by butyrate with induction of GADD153 in HCT116 cells. In the RKO CRC cell line, butyrate activated the c-Jun N-terminal kinase (JNK) but not the p38 arm of the MAPK signalling pathway and this correlated with caspase activation and apoptosis(Reference Zhang, Zhou and Bao46). Tong et al. (Reference Tong, Yin and Song45) reported the loss of expression in both mouse and human intestinal tumours, lending further support to its role in intestinal tumour formation. Butyrate is also reported to increase phosphorylation of p38 and its downstream target heat shock protein 27 in both HCT116 CRC cells and MCF7 breast cancer cells(Reference Fung, Brierley and Henderson29, Reference Yonezawa, Kobayashi and Obara47). Although the apoptotic response to butyrate is dependent on the cell line being studied, in each case, the activation of MAPK signalling occurs within minutes of butyrate exposure. This indicates that induction of a cell stress response occurs as an early event in butyrate-induced apoptosis.
Effect of butyrate on processes involved in tumorigenesis
Genomic instability and epigenetic regulation
Histone deacetylase inhibition
Regulation of gene expression via inhibition of HDAC activity is the primary mechanism associated with butyrate-induced apoptosis. HDAC inhibitors (including butyrate) target the transcription of less than 10 % of the human genome selectively and their cellular effects are also mediated by modulating the acetylation state of both histone and non-histone proteins including transcription factors, structural proteins and proteins involved in signal transduction(Reference Wilson, Chueh and Togel48). Our knowledge of the number of non-histone proteins modified as a result of butyrate treatment is limited but is known to include the Sp1 and Sp3 transcription factors(Reference Waby, Chirakkal and Yu49, Reference White, Mulligan and King50). Although the functional consequences of Sp1 and Sp3 acetylation by butyrate are not known, acetylation of Sp1 has been hypothesised to increase p21 expression and mediate p53-dependent cell cycle arrest in CRC cell lines(Reference Waby, Chirakkal and Yu49). Identification of other non-histone protein targets acetylated by butyrate treatment may provide new insights into butyrate's mechanisms of action.
Role of p53 in cell cycle arrest and apoptosis
In vitro studies have shown consistently that butyrate induces cell cycle arrest and apoptosis in both a p53-dependent and -independent manner at physiologically relevant concentrations (0·6–5 mmol/l)(Reference Janson, Brandner and Siegel51). In CRC cell lines, butyrate down-regulates the expression of p53 mRNA and protein and also directly increases the expression of p53 target genes (e.g. p21WAF1, p27 and cyclin-dependent kinases) to induce cell cycle arrest(Reference Gope and Gope52, Reference Nakano, Mizuno and Sowa53). Activation of p53 and its translocation to the nucleus is regulated by post-translational modifications, including acetylation by histone acetyltransferases such as p300, that can increase both the stability and pro-apoptotic activity of p53(Reference Yuan, Huang and Ishiko54). Prolonged or transient hyper-acetylation of p53 by HDAC inhibitors such as butyrate may represent an additional mechanism making an impact on p53-dependent apoptosis(Reference Terui, Murakami and Takimoto55).
Butyrate and micro-RNA regulation
Butyrate also alters gene expression independently of HDAC inhibition by regulating the expression of micro-RNA (miRNA)(Reference Hu, Dong and Dalal56, Reference Zhang, Li and Nan57). micro-RNA are non-protein coding RNA species that regulate translation of their respective mRNA targets. Over 1000 human miRNA have been identified (www.miBase.org accessed June 2011). Despite the intensity of effort, only a small number of miRNA, notably miR-31 and those of the miR-194/-215 and miR-143/-145 clusters, have consistently been associated with colorectal tumorigenesis(Reference Necela, Carr and Asmann58). In vitro studies have demonstrated that butyrate regulates the expression of miRNA in HCT116 cells and in human CRC stem cells characterised by CD133 cell surface expression when compared to respective control cells, including those mentioned previously(Reference Hu, Dong and Dalal56, Reference Zhang, Li and Nan57). Establishing the role of miRNA in the regulation of gene transcription is still in its infancy and the true potential that butyrate may have on miRNA expression, or regulation of its function, remains to be elucidated.
Inflammation and the immune response
SCFA have anti-inflammatory effects in the large bowel. In patients with distal ulcerative colitis, rectal administration of either SCFA mixtures(Reference Breuer, Buto and Christ59, Reference Breuer, Soergel and Lashner60) or butyrate alone(Reference Scheppach, Sommer and Kirchner61) has been shown to be effective at ameliorating the clinical symptoms of the disease. In intestinal epithelial cells, butyrate modulates colonic inflammation by reducing the expression of IL-8(Reference Huang, Katz and Martin62) and inhibiting inducible NO synthase expression(Reference Stempelj, Kedinger and Augenlicht63). Butyrate also alleviates oxidative stress and protects against oxidative DNA damage in cultured CRC cells and in colonic mucosal cells(Reference Rosignoli, Fabiani and De Bartolomeo64, Reference Sauer, Richter and Pool-Zobel65). This is supported further by reports of altered expression of proteins involved in free-radical scavenging and elevated activity of glutathione S-transferase, a protein responsible for metabolising potential carcinogens(Reference Fung, Lewanowitsch and Henderson30, Reference Ebert, Klinder and Peters66). Furthermore, incubation of HT29 cells with 4 mmol/l butyrate suppresses cyclo-oxygenase-2 expression and activity(Reference Tong, Yin and Giardina67). However, data derived from in vivo experiments where colorectal tumour tissue was exposed to butyrate have been inconclusive, possibly due to the size of the patient cohorts tested, tumour tissue heterogeneity and potential confounding by clinical parameters(Reference Jahns, Wilhelm and Jablonowski68). This is a critical factor in validating the relevance of data derived from model systems to human CRC and it is highly desirable to invest in such an effort.
Activation of NF-κB is one of the primary contributors to the development of inflammation-associated carcinogenesis, including CRC arising from chronic inflammatory conditions such as ulcerative colitis(Reference Greten, Eckmann and Greten69). Although butyrate regulates the activity of NF-κB, the signalling mechanisms involved in this process are not known. Constitutive activation of NF-κB has been reported in approximately 40 % of colorectal tumour tissues(Reference Sakamoto, Maeda and Hikiba70) and NF-κB activation in tumour cells promotes survival both by potentiating the inflammatory response through the activation of signalling pathways regulated by pro-inflammatory cytokines and by regulating the expression of anti-apoptotic genes(Reference Chen, Edelstein and Gelinas71, Reference Zong, Edelstein and Chen72).
Role of the SLC5A8 transporter in the inflammatory response and colorectal cancer
The Na-linked solute transporter SLC5A8 (solute carrier family 5, member 8) is silenced epigenetically in a number of different cancers(Reference Li, Myeroff and Smiraglia73). This transporter is expressed in the colon, kidney and thyroid and recognises (and transports) monocarboxylic acids, including butyrate. Methylation of the SLC5A8 gene and its subsequent loss of expression have been detected in 59 % of CRC adenoma and tumour tissues(Reference Li, Myeroff and Smiraglia73). Silencing of SLC5A8 in CRC has been associated with mutant and inactive adenomatous polyposis coli (APC) protein and aberrant Wnt signalling(Reference Thangaraju, Cresci and Itagaki74). In vitro studies have shown that butyrate transport by SLC5A8 inhibits HDAC activity and the growth of tumour cells(Reference Thangaraju, Cresci and Itagaki74, Reference Thangaraju, Gopal and Martin75). Thangaraju et al. (Reference Thangaraju, Cresci and Itagaki74) also reported that loss of SLC5A8 expression on CRC cell lines was inversely linked with apoptosis, and that apoptosis occurred as a result of inhibition of HDAC activity and activation of the caspase cascade by butyrate.
Involvement of SCFA receptors in the inflammatory response and colorectal cancer
SCFA augment the immune and inflammatory response by influencing immune cell functions such as chemotaxis, phagocytosis, reactive oxygen species production and cytokine/chemokine release. Butyrate reduces reactive oxygen species production and cytokine release in activated neutrophils (at approximately 1·6 mmol/l)(Reference Vinolo, Rodrigues and Hatanaka76) and plays a role in immune cell migration in vivo (Reference Bailon, Cueto-Sola and Utrilla77, Reference Vinolo, Rodrigues and Hatanaka78). Recently, production of acetate by bifidobacteria was found to improve the immune defence function of intestinal epithelial cells in vivo (Reference Fukuda, Toh and Hase79). These effects have been attributed, in part, to the activation of G-protein coupled receptors (GPR), in particular GPR109A and GPR43. The GPR40 family comprises receptors for SCFA and medium-chain fatty acids, with GPR41 and GPR43 having the highest affinity for SCFA ( < 5 carbons)(Reference Brown, Goldsworthy and Barnes80–Reference Nilsson, Kotarsky and Owman82). Although these receptors display millimolar affinity for the SCFA, these concentrations are readily achievable in the colon. The role of GPR43 in inflammatory conditions has been studied widely using isolated immune cells and in mouse models of intestinal inflammation(Reference Le Poul, Loison and Struyf81, Reference Aoyama, Kotani and Usami83–Reference Sina, Gavrilova and Forster85). In mouse models of both acute and chronic colitis induced by dextran sulphate sodium, absence of GPR43 expression resulted in greater colonic inflammation and compromised mucosal integrity when compared to wild-type littermates expressing the receptor(Reference Maslowski, Vieira and Ng84). Consumption of acetate in the drinking water resulted in improved inflammatory indices in wild-type mice but not in GPR43 knockout mice, indicating that activation of this receptor by SCFA plays a role in modulating the inflammatory response. Conversely, Sina et al. (Reference Sina, Gavrilova and Forster85) reported that loss of GPR43 expression reduced colonic inflammation in the chronic dextran sulphate sodium mouse model of colitis. These apparently contradictory results may be attributed to differences in the dose and duration of dextran sulphate sodium administration between these two studies. Nevertheless, both studies report that stimulation of GPR43 by SCFA (acetate(Reference Maslowski, Vieira and Ng84) and propionate and butyrate(Reference Sina, Gavrilova and Forster85)) is essential for immune cell recruitment and that this chemotactic response is mediated by the MAPK pathway.
GPR43 has been implicated in CRC prevention where it may have a tumour-suppressive role(Reference Tang, Chen and Jiang86). Loss of GPR43 expression occurs in colorectal adenocarcinoma tissue when compared to normal mucosa and reduced expression has been noted in colorectal hyperplasia and benign colorectal disease including polyps(Reference Tang, Chen and Jiang86). In a panel of nine CRC cell lines, Tang et al. (Reference Tang, Chen and Jiang86) determined that GPR43 was expressed in the HT29 CRC cell line only. They established also that re-expression of this receptor in CRC cells and activation by either butyrate (50% inhibitory concentration (IC50) 0·8 mmol/l) or propionate (IC50 2 mmol/l) inhibited proliferation and induced apoptosis and cell cycle arrest, providing further support for the role of GPR43 in maintaining normal cellular function. This effect of propionate is important, as it has been reported to induce some effects which are similar to butyrate, albeit at much higher concentrations. GPR43 expression has been studied in the MCF7 breast cancer cell line where its activation by each of the SCFA (at 10 mmol/l) induced a cell stress response mediated specifically by p38 MAPK signalling(Reference Yonezawa, Kobayashi and Obara47). These data indicate that GPR43 plays a pivotal role in activating the signalling pathways associated with the reported cellular and anti-tumorigenic effects of butyrate (and other SCFA). Despite this, GPR43-mediated cell signalling events in both the normal colon, colonic inflammatory conditions and in the development of CRC need to be determined.
GPR109A was initially identified as the receptor for nicotinic acid(Reference Soga, Kamohara and Takasaki87–Reference Wise, Foord and Fraser89) and belongs to a receptor subfamily that includes GPR81 and GPR109B(Reference Ahmed, Tunaru and Offermanns90). Although GPR109A and GPR109B are similar structurally and have a similar expression pattern, they differ in their ligand specificity(Reference Wise, Foord and Fraser89, Reference Taggart, Kero and Gan91). GPR109A is activated by nicotinic acid, 3-hydroxybutyrate and butyrate, whereas GPR109B displays much low affinity for them. Butyrate binds and activates the GPR109A receptor (50% effective concentration (EC50) 1·6 mmol/l), but has no effect on GPR109B and neither receptor displays affinity for acetate or propionate(Reference Taggart, Kero and Gan91). GPR109A is expressed in the human colon and the expression of this receptor is dysregulated in CRC(Reference Thangaraju, Cresci and Liu92). A similar expression pattern was also determined in a mouse model of CRC and in a panel of human CRC cell lines. The authors determined also that silencing of GPR109A in CRC occurs as a result of DNA methylation. Subsequent re-expression of GPR109A in CRC cell lines and its activation by 1 mmol/l butyrate abolished NF-κB activation and induced apoptosis. Apoptosis occurred independently of HDAC inhibition, lending further support to the existence of alternate mechanisms involved in butyrate-induced apoptosis. Although GPR109A is also expressed on immune and inflammatory cells, its role in these cells has yet to be determined.
Cancer cell metabolism
Tumour cells display an altered cellular metabolism that can be viewed as an adaptive response to an hypoxic microenvironment or as a result of mutations in oncogenes and tumour-suppressor genes leading to higher glycolytic activity and enhanced energy production – a phenomenon known as the ‘Warburg effect’(Reference Warburg93). Although this characteristic of cancer cells has been studied widely, the mechanisms linking this switch in cell metabolism from a normal phenotype to cancer cell survival are uncertain. Transcription factors such as hypoxia inducible factor (HIF), tumour protein p53, octamer-binding protein 1 (OCT1), NF-KB and avian myelocytomatosis viral oncogene homolog (MYC) have been linked to dysregulated expression of nutrient transporters, glycolytic enzymes, and proteins involved in mitochondrial function(Reference Cairns, Harris and Mak94) and it is possible that butyrate may target these transcription factors to alter cell metabolism. In vitro studies and those involving isolated human colonocytes have indicated that butyrate modulates the levels and activity of transcription factors. In the case of c-myc, 5 mmol/l butyrate inhibits its transcription and protein expression in malignant cells, possibly via its ability to inhibit HDAC activity(Reference Emenaker and Basson95, Reference Wilson, Velcich and Arango96). In vitro studies also demonstrate that, while butyrate increases transcription of the HIF-1α gene, it represses the transcriptional regulatory activity of HIF-1α protein by inhibiting its translocation to the nucleus in Caco2 cells(Reference Pellizzaro, Coradini and Daidone97, Reference Zgouras, Wachtershauser and Frings98). These studies demonstrate further that inhibition of VEGF expression and angiogenesis occurs with butyrate incubation, but the effects on cellular metabolism were not investigated. Regulation of HIF-1α translocation by butyrate presents a potential mechanism contributing to its effects on angiogenesis.
Regulation of cancer cell metabolism by butyrate
Recent studies have shown that CRC cells, which are highly dependent on glycolysis, can acquire a unique ability to utilise both butyrate and glucose as their energy source and that this metabolic switch can be induced by the former(Reference Serpa, Caiado and Carvalho36). This capacity for both butyrate and glucose metabolism by colorectal tumour cells is supported by an elevated expression of solute transporters with high affinity for either substrate. In particular, monocarboxylate transporter 1 (MCT1, SLC16A1 (solute carrier family 16, member 1)) and GLUT type 1 (GLUT1, SLC2A1 (solute carrier family 2, member 1)) are elevated in CRC tissue when compared to adjacent normal mucosa(Reference Koukourakis, Giatromanolaki and Harris99, Reference Pinheiro, Longatto-Filho and Scapulatempo100). MCT1 is expressed widely in many different cell types and has been characterised as the primary butyrate transporter in the colon. It is expressed in healthy colon tissue and in many different cultured CRC cell lines(Reference Andriamihaja, Chaumontet and Tome101, Reference Tamai, Takanaga and Maeda102). Reports on the expression of MCT1 in CRC tissue have been conflicting with reported increases(Reference Koukourakis, Giatromanolaki and Harris99, Reference Pinheiro, Longatto-Filho and Scapulatempo100) or decreases(Reference Lambert, Wood and Ellis103) in expression. In addition to facilitating butyrate entry into the cell, MCT1 functions to remove lactate, a potentially cytotoxic metabolic by-product of glycolysis, indicating that it plays a dual role to enhance the survival of CRC cells(Reference Koukourakis, Giatromanolaki and Harris99). Butyrate also increases the expression of MCT1 on CRC cells in a dose-dependent manner and this is partially attributed to NF-κB activity(Reference Borthakur, Saksena and Gill104).
GLUT1 is expressed widely and its expression is elevated in cells from many different types of cancer. Its level of expression in tumours, including CRC, has been correlated with poor clinical outcomes(Reference Haber, Rathan and Weiser105). The mechanisms involved in the regulation of GLUT1 expression in cancer cells are not completely understood. Induction of its expression has been linked with an hypoxic microenvironment and regulation by HIF-1. However, there are alternative mechanisms(Reference Kim, Gao and Dang106). Using a panel of CRC cell lines, Yun et al. (Reference Yun, Rago and Cheong107) recently showed that increased GLUT1 expression is independent of HIF-1 activity and may instead be a downstream consequence of dysregulated signalling pathways caused by mutations in the KRAS or BRAF (v-raf murine sarcoma viral oncogene homolog B1) genes. It is not known if butyrate is able to regulate the expression of GLUT1 in CRC cells. Although these findings are contradictory, they can be reconciled if this altered metabolic profile is due to the presence of a butyrate-resistant sub-population of CRC cells. These cells would not succumb to apoptosis with butyrate exposure, but could proliferate to establish tumours. In the Caco2 CRC cell line, butyrate inhibits the pyruvate dehydrogenase complex, reducing the capacity for glycolysis and inducing a switch to a butyrate-oxidising phenotype providing a potential explanation for the development of a butyrate-resistant cell population(Reference Blouin, Penot and Collinet108). This is an established feature of the regulation of pyruvate dehydrogenase in a number of tissues. Glutamine metabolism was increased to favour the production of precursors for fatty acid synthesis, a process essential for cell proliferation. The group further demonstrated that this metabolic switch occurred as a result of HDAC inhibition by butyrate.
Recently, a link was established between HIF-1α and APC, further supporting the role of HIF-1α in the early stages of carcinogenesis(Reference Newton, Kenneth and Appleton109). In this study, HIF-1α was found to bind directly to the promoter region of APC, inhibiting its expression and (potentially) enhancing tumour cell survival(Reference Newton, Kenneth and Appleton109). This study also showed that depletion of wild-type APC protein, but not a mutant one, resulted in elevated HIF-1α activity. Mutations in APC are common to CRC, and thus up-regulation of HIF-1α in these cells may provide a further competitive advantage to tumour cells by altering their metabolism to promote adaptation to an hypoxic environment. Limitations in O2 supply could be an important factor in CRC development and also help to explain some of the unanswered questions in the aetiology of the disease. Cigarette smoking is an established risk factor for CRC and heavy use leads to the significant accumulation of erythrocyte carbonmonoxyhaemoglobin(Reference Williamson, Pols and Illman110). Blood carbonmonoxyhameoglobin leads to substantial changes in metabolic processes (e.g. ethanol metabolism(Reference Topping, Fishlock and Trimble111) and lipoprotein catabolism(Reference Gardner, Topping and Mayes112)) through decreased tissue O2 consumption. In contrast, greater SCFA production (through fermentation) leads to greater visceral perfusion and, hence, increased O2 supply although infusion studies suggest that the effect of butyrate is rather less than the other SCFA(Reference Topping and Clifton12).
Butyrate and butyrate analogues as chemotherapeutic agents
HDAC inhibitors are being investigated seriously as potential chemotherapeutic agents. Suberoylanilide hydroxamic acid was recently approved for the treatment of cutaneous T-cell lymphoma(Reference Mann, Johnson and Cohen113). Experiments with various cancer cell lines have been conducted to determine if combined treatment with DNA methyltransferase inhibitors (e.g. 5-aza-2′-deoxycytidine) and prototype HDAC inhibitors (e.g. trichostatin A, butyrate) could restore the function of key tumour-suppressor genes(Reference Fang, Chen and Lu114). Studies such as these are aimed primarily at determining the potential efficacy of combined treatments for cancer therapy. Butyrate itself is disqualified as a candidate drug on several counts. These include its ready uptake and metabolism by many cell types in the body, leading to rapid clearance and short half-life in the circulation. To overcome these limitations, many studies have explored the potential of butyrate derivatives, with 4-phenylbutyrate and tributyrin being the most promising(Reference Kang, Lee and Lee115–Reference Miyoshi, Sakaki and Usami118). In vitro studies comparing the structural analogues of butyrate have shown that the apoptotic properties of butyrate are dependent on the lack of substitution at the 2- and 3-positions of the carboxylate backbone(Reference Ooi, Good and Williams119, Reference Ooi, Good and Williams120). Furthermore, these studies identified 4-benzoylbutyrate and 4-phenylbutyrate to be the most potent of the thirty-two analogues compared, indicating that a three-atom spacer between any bulky moiety and carboxyl group may also be essential for the anti-tumorigenic properties of any butyrate-based pharmaceutical. Although these studies did not identify analogues that were more potent than butyrate in vitro, they provide a starting point for the development of novel therapeutic agents.
Conclusion and future perspectives
CRC is a disease where there is clear potential for lowering the risk through dietary and lifestyle changes, with 30–60 % of tumours being preventable. The evidence available suggests that the consumption of diets high in fibre, in particular RS, and low in fats and proteins is protective against CRC development. The role of fibre fermentation is an area of growing significance in disease aetiology, with increasing attention being given to SCFA(Reference Cassidy, Bingham and Cummings121). It appears that SCFA mediate many of the effects previously ascribed to fibre alone. Butyrate is central to the proposed link between diet and protection against CRC. There appears to be a clear pathway that integrates, at a population level, the beneficial effects of a high-fibre diet (especially fermentable fibre), the formation of SCFA by colonic microbiota and demonstration of a role for the SCFA in maintaining physiological function in the colon including cell growth and regulation. Key to this hypothesis is the ability of butyrate to influence cellular processes to support a normal cell population and induce apoptosis and inhibit tumorigenesis (Fig. 2). While in vitro experiments indicate that HDAC inhibition is the primary mechanism for butyrate's anti-tumorigenic effects, the evidence for additional cellular pathways contributing to butyrate's pro-apoptotic and anti-proliferative activities in tumour cells is mounting. The current body of knowledge offers promise for containing CRC, but it is a clear priority to delineate these mechanisms in experimental systems and move on to human population and intervention studies to determine their true potential for risk reduction. Nutritional trials in free-living subjects have shown that it is possible to raise faecal SCFA (including butyrate) to levels found in low-risk populations such as native Africans consuming traditional foods(Reference Bird, Vuaran and King122, Reference McOrist, Miller and Bird123). Although these studies demonstrate that such interventions are feasible, it appears that some individuals do not show the expected rise in SCFA on increasing RS consumption(Reference McOrist, Miller and Bird123). This may reflect differences in the capacity of their large-bowel microbiome to ferment starch(Reference Cummings, Beatty and Kingman124). Clearly, it is imperative to show that any increase in colonic butyrate supply can be effective and sustained.
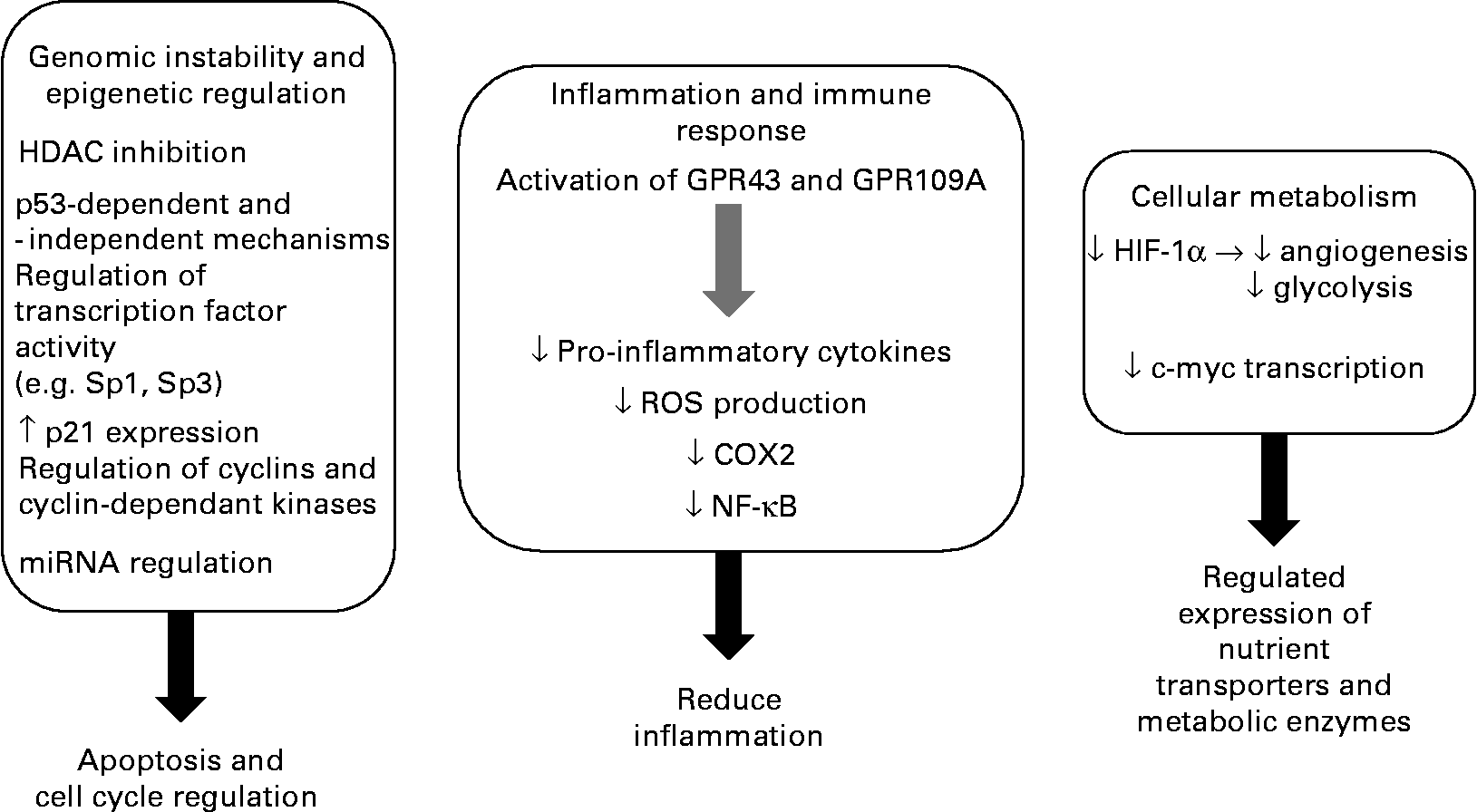
Fig. 2 Summary of the anti-tumorigenic effects of butyrate. HDAC, histone deacetylase; miRNA, micro-RNA; GPR43, G-protein coupled receptor 43; GPR109A, G-protein coupled receptor 109A; ROS, reactive oxygen species; COX2, cyclo-oxygenase-2; HIF-1α, hypoxia inducible factor.
Acknowledgements
The authors would like to thank Dr Leanne Purins and Dr Julie Clarke for helpful discussions. The present work was supported by the CSIRO Preventative Health and Food Futures National Research Flagships. The authors declare no conflicts of interest. K. Y. C. F., D. L. T. and R. H. wrote the manuscript. K. Y. C. F., L. C., T. L., R. H. and D. L. T. contributed to the ideas and participated in the editing. All authors read and approved the final manuscript.



