



Introduction
Based on comparative morphology, the eucestodes Tetraphyllidea, Lecanicephalidea, Onchoproteocephalidea (syn. Proteocephalidea), Nippotaeniidea, Tetrabothriidea and Cyclophyllidea are closely related taxa (Hoberg et al., Reference Hoberg, Mariaux, Justine, Brooks and Weekes1997). Evidence from 28S recombinant DNA (rDNA) and 18S rDNA (Olson et al., Reference Olson, Littlewood, Bray and Mariaux2001, Reference Olson, Poddubnaya, Littlewood and Scholz2008; Waeschenbach et al., Reference Waeschenbach, Webster, Bray and Littlewood2007; Caira et al., Reference Caira, Jensen, Waeschenbach, Olson and Littlewood2014) and the large fragments of mitochondrial DNA (mtDNA) (Waeschenbach et al., Reference Waeschenbach, Webster and Littlewood2012) suggest that the acetabulate lineages (Litobothriidea, Lecanicephalidea, Nippotaeniidea, Cyclophyllidea and Tetrabothriidea) form a monophyletic group. Owing to the non-monophyly of the order Tetraphyllidea, the new order Rhinebothriidea was established to house the tetraphyllideans with stalked acetabula (Healy et al., Reference Healy, Caira, Jensen, Webster and Littlewood2009), and the new order Onchoproteocephalidea was established for ten described genera of hook-bearing tetraphyllideans and the members of the order Proteocephalidea (Caira et al., Reference Caira, Jensen, Waeschenbach, Olson and Littlewood2014).
Onchoproteocephalidean tapeworms, with a cosmopolitan distribution, represent a diverse group of parasites with 316 valid species in bony fish, lizard, snake and amphibian hosts (Caira et al., Reference Caira, Jensen and Ivanov2017), and 246 valid species in elasmobranch hosts (Caira et al., Reference Caira, Jensen and Ivanov2017). The traditionally accepted families Proteocephalidae and Monticelliidae have been abandoned, and the only family Protecephalidae has been split into a number of subfamilies and genera (de Chambrier et al., Reference de Chambrier, Coquille, Mariaux and Tkach2009). 28S rDNA-based phylogeny suggests that most of the presently recognized subfamilies (and genera) appear to be non-monophyletic, and a deep systematic reorganization of the order is thus needed (de Chambrier et al., Reference de Chambrier, Waeschenbach, Fisseha, Scholz and Mariaux2015). Regardless of the validity of subfamilies, the Gangesiinae and the Acanthotaeniinae appear to be the most primitive, and the Old World (Palaearctic Region) origin of onchoproteocephalideans in freshwater fish is confirmed by the phylogenetic analysis of the 28S rDNA sequence (de Chambrier et al., Reference de Chambrier, Zehnder, Vaucher and Mariaux2004, Reference de Chambrier, Coquille, Mariaux and Tkach2009, Reference de Chambrier, Waeschenbach, Fisseha, Scholz and Mariaux2015) and the internal transcripted spacer sequence and 18S rRNA (Hypša et al., Reference Hypša, Škerikoa and Scholz2005). Owing to the non-monophyly of subfamilies and the polytomy of the phylogenetic tree of siluriform parasites from the Neotropics, large mtDNA fragments and multiple genes are obviously needed (de Chambrier et al., Reference de Chambrier, Waeschenbach, Fisseha, Scholz and Mariaux2015).
Owing to its maternal inheritance, a lack of recombination and a fast rate of evolution, the haploid mitochondrial genome has proven to be a useful marker for population studies, species identification and phylogenetics (Huyse et al., Reference Huyse, Buchmann and Littlewood2008). Using gene sequences and gene arrangements from the complete mitochondrial genome, the phylogenies of some parasitic platyhelminths have been reconstructed (Littlewood et al., Reference Littlewood, Lockyer, Webster, Johnston and Le2006). Although mitogenomes for more than 40 cyclophyllideans are available in GenBank, the only onchoproteocephalidean mitogenome available in GenBank is that of Testudotaenia sp. WL-2016, belonging to the newly erected subfamily Testudotaeniinae (de Chambrier et al., Reference de Chambrier, Coquille, Mariaux and Tkach2009).
Gangesia oligonchis (Gangesiinae) parasitizes in the intestine of the bullhead catfish, Tachysurus fulvidraco (Siluriformes: Bagridae), distributed in Russia (Ash et al., Reference Ash, de Chambrier, Shimazu, Ermolenko and Scholz2015) and China (Fu et al., Reference Fu, Li, Zou, Zhang, Wu, Li, Wang and Xi2019). Thus the mitogenome of G. oligonchis was sequenced and characterized to provide mitogenomic data for future studies on inter-relationships of onchoproteocephalideans.
Materials and methods
Specimen collection and DNA extraction
Tapeworms were collected from the intestine of the bullhead catfish (T. fulvidraco), which was anesthetized with 0.02% MS-222, from Liangzi Lake in Hubei Province, China (30°11′05″N, 114°37′34″E). Their identity with Gangesia oligonchis was confirmed using morphology and partial 28S rDNA data (Fu et al., Reference Fu, Li, Zou, Zhang, Wu, Li, Wang and Xi2019). Voucher specimen (accession number: IHB-Gangesia001) was deposited in the museum at the Institute of Hydrobiology, Wuhan, China.
Tapeworm specimens were preserved in 100% ethanol and stored at 4 °C. Total genomic DNA was extracted from a single worm using a TIANamp Micro DNA Kit (Tiangen Biotech, Beijing, China) according to the manufacturer's protocol, and stored at −20 °C.
PCR and DNA sequencing
Sequences from GenBank were used to design five primer pairs (see supplementary material table S1). These primers were used to amplify partial sequences of the rrnS, cox1, cytb, nad2 and nad4 genes. Based on these fragments, seven specific primers were designed for subsequent PCR amplification. PCR reactions were conducted in a 20 µl reaction mixture, containing 7.4 µl dd H2O, 10 µl 2×PCR buffer (Mg2+, dNTP plus, Takara), 0.6 µl of each primer, 0.4 µl rTaq polymerase (250 U, Takara, Dalian, China) and 1 µl DNA template. Amplification was performed under the following conditions: initial denaturation for 2 min at 98 °C, followed by 40 cycles: 10 s at 98 °C, 15 s at 48–60 °C, 1 min at 68 °C and a final extension for 10 min at 68 °C. PCR products were sequenced bidirectionally at Sangon Company (Shanghai, China) using the primer walking strategy.
Sequence annotation analyses
After checking the quality of the sequences, amplification of the complete mitochondrial genomic sequence of G. oligonchis was assembled using the DNAstar program (Burland, Reference Burland2000) and confirmed by BLAST (Altschul et al., Reference Altschul, Gish, Miller, Myers and Lipman1990). Mitogenome annotation followed the procedure described by Li et al. (Reference Li, Zhang, Boyce, Xi, Zou, Wu, Li and Wang2017, Reference Li, Fu, Zhang, Boyce, Xi, Zou, Li, Wu and Wang2018). Protein-coding genes (PCGs) were detected by searching for open reading frames (employing genetic code 9, invertebrate mitochondrial) and by checking the nucleotide alignments against homologues. All of the transfer RNAs (tRNAs) were predicted and confirmed using the ARWEN program (Laslett & Canback, Reference Laslett and Canback2008) and MITOS web server (Bernt et al., Reference Bernt, Donath, Juhling, Externbrink, Florentz, Fritzsch, Putz, Middendorf and Stadler2013). Similarly, the positions of rrnL and rrnS were preliminarily located using the MITOS program (Bernt et al., Reference Bernt, Donath, Juhling, Externbrink, Florentz, Fritzsch, Putz, Middendorf and Stadler2013), and their ends were assumed to extend to the boundaries of their flanking genes. Tandem repeats in the non-coding region were identified using the Tandem Repeats Finder program (Benson, Reference Benson1999), and the secondary structure was predicted using Mfold software (Zuker, Reference Zuker2003). Codon usage and relative synonymous codon usage (RSCU) of the 12 PCGs were computed and sorted using the PhyloSuite program (Zhang et al., Reference Zhang, Gao, Li, Jakovlić, Zou, Zhang and Wang2018), and the RSCU figures were finally drawn using ggplot2 plugin (Hadley, Reference Hadley2009). The circular map of the G. oligonchis mitogenome was drawn with the mitochondrial visualization tool MTVIZ (http://pacosy.informatik.uni-leipzig.de/mtviz/).
Phylogenetic analyses
Phylogenetic analyses were carried out using the newly sequenced mitogenome of G. oligonchis and the 35 cestode mitogenomes available in GenBank (table 1 and supplementary material table S2). Two species of trematode, Dicrocoelium dendriticum (Rudolphi, 1819) (NC 025280) and Dicrocoelium chinensis (Sudarikov and Ryjikov, 1951) (NC 025279), were used as outgroups. The PhyloSuite program was used to generate the AT content and the GC skew (see supplementary material table S2) and the *.sqn file for GenBank submission. A Fasta file with the nucleotide sequences for all 36 genes (12 PCGs, 2 rRNAs and 22 tRNAs) for the 35 cestodes was downloaded from GenBank using PhyloSuite. All genes were aligned with the MAFFT program (Katoh & Standley, Reference Katoh and Standley2013) integrated in PhyloSuite, wherein codon-alignment mode was used for the 12 PCGs, and normal-alignment mode was used for the remaining RNAs (two rRNAs and 22 tRNAs). PhyloSuite was then used to concatenate these alignments and generate input files for the phylogenetic analyses, conducted using maximum likelihood (ML) and Bayesian inference (BI) methods. The most appropriate evolutionary models for the dataset were determined using ModelGenerator v0.8527 (Keane et al., Reference Keane, Creevey, Pentony, Naughton and McInerney2006). Based on the Akaike information criterion, GTR + I + G was chosen as the optimal model of nucleotide evolution. ML analysis was performed using the RaxML GUI (Silvestro & Michalak, Reference Silvestro and Michalak2011) and the ML + rapid bootstrap (BP) algorithm with 1000 replicates. BI analysis was performed with MrBayes 3.2.1 (Ronquist et al., Reference Ronquist, Teslenko and van der Mark2012) with default settings, and 6 × 106 metropolis-coupled Markov chain Monte Carlo generations. Bayesian posterior probability (BPP) values were calculated in a consensus tree after discarding the first 25% samples as burn-in.
Table 1. The list of cestode species used for comparative analyses mitogenomes.

Results
Genome organization and base composition
The typical circular duplex molecule mitogenome of G. oligonchis was 13,958 bp in length (GenBank accession number: MF314173). Apart from lacking the Atp8 gene, which is typical of parasitic flatworms (Le et al., Reference Le, Blair and McManus2002), the mitogenome contained the standard 36 genes: 22 tRNA genes, two rRNA genes and 12 PCGs, as well as two major non-coding regions (mNCRs) (fig. 1). All genes were transcribed from the same strand. Five overlapping regions were found in the genome (table 2).
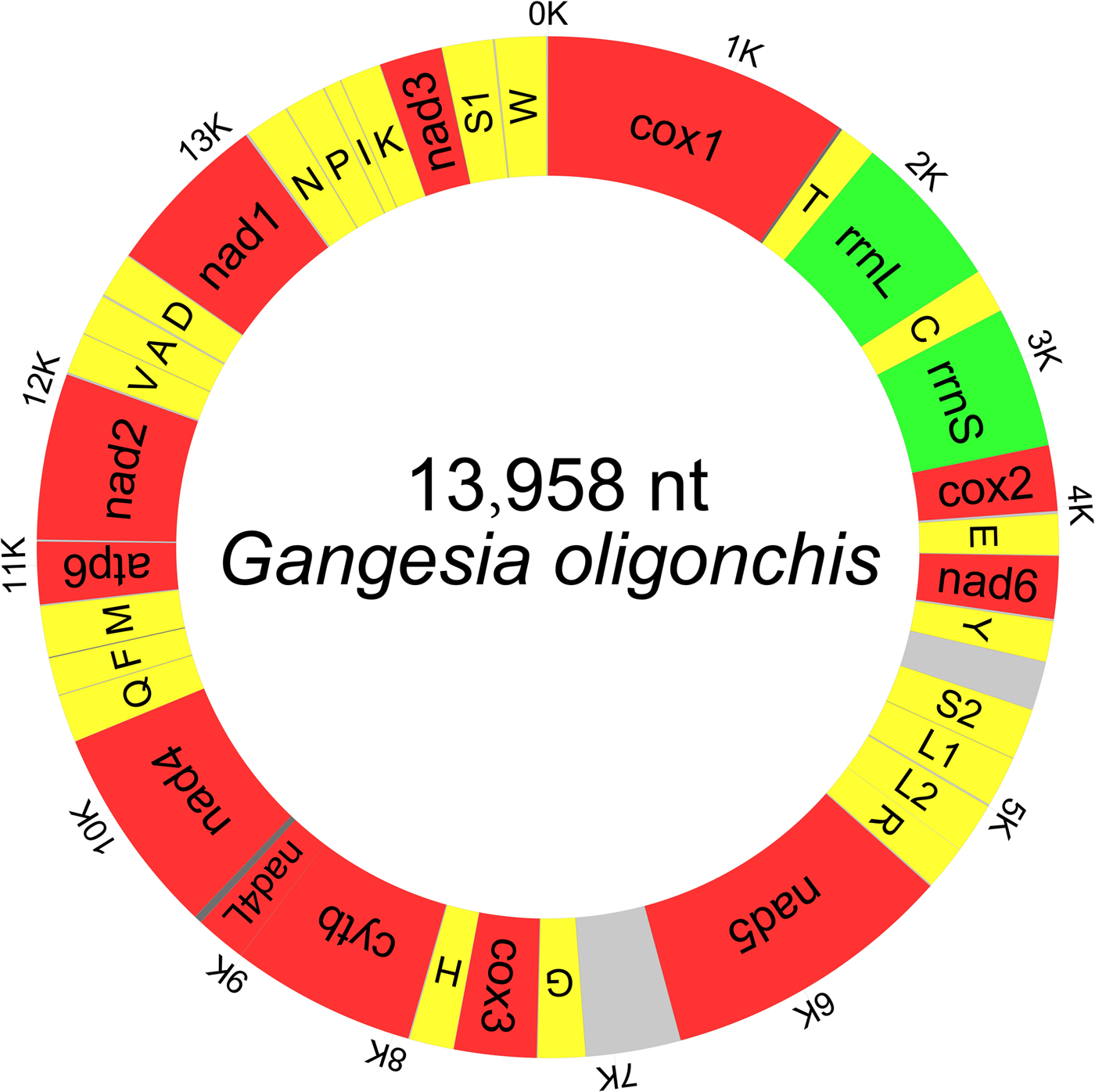
Fig. 1. Map of the complete mitochondrial genome of Gangesia oligonchis. All 36 genes and major non-coding regions are displayed.
Table 2. The organization of the mitochondrial genome of Gangesia oligonchis.

An A + T bias was detected in the mitogenome of G. oligonchis (A = 23.0%, T = 43.3%, C = 12.9%, G = 20.8%). The nucleotide composition of the complete mitogenome of G. oligonchis was strongly skewed away from A, in favour of T, and was biased towards G (AT skew = −0.307, GC skew = 0.235) (table 3). The mitogenomes of G. oligonchis and Testudotaenia sp. WL-2016 shared 71.1% nucleotide identity, with 65.4–78.5% identity in PCGs and rRNA genes (table 2).
Table 3. Nucleotide composition of the protein-coding genes, tRNAs, rRNAs and non-coding region of mitochondrial genomes of Gangesia oligonchis and Testudotaenia sp. WL-2016.

Protein-coding genes and codon usage
The total length of the concatenated 12 PCGs was 10,122 bp, with the average A + T content of 65.4%, ranging from 63.8% (cox2) to 71% (nad3) (table 3). The start codon ATG was commonly found in ten PCGs, whereas the start codon GTG was most commonly found in cox3 and nad6. The most frequent stop codons were TAG (for eight PCGs), followed by TAA (four PCGs). Codon usage, relative synonymous codon usage (RSCU), and codon family proportion (corresponding to the amino acid usage) of these two onchoproteocephalideans are presented in fig. 2. Leucine (14.9%), phenylalanine (12.9%), valine (10.4%) and isoleucine (6.7%) were the most frequent amino acids in the PCGs of G. oligonchis, which is also observed in Testudotaenia sp. WL-2016 (fig. 2). In particular, all second codon positions of the codons encoding these amino acids were T, corresponding to its relatively high T skewness (AT skew = −0.465, table 3). The most frequent codons were TTT (phenylalanine, 11.3%) and TTA (leucine, 6.4%), both consisted of A and T. Codons ending in A or T were predominant (blue and green bar in fig. 2), which corresponds to the high A + T content of the third coding position of all PCGs in G. oligonchis (67.7%).
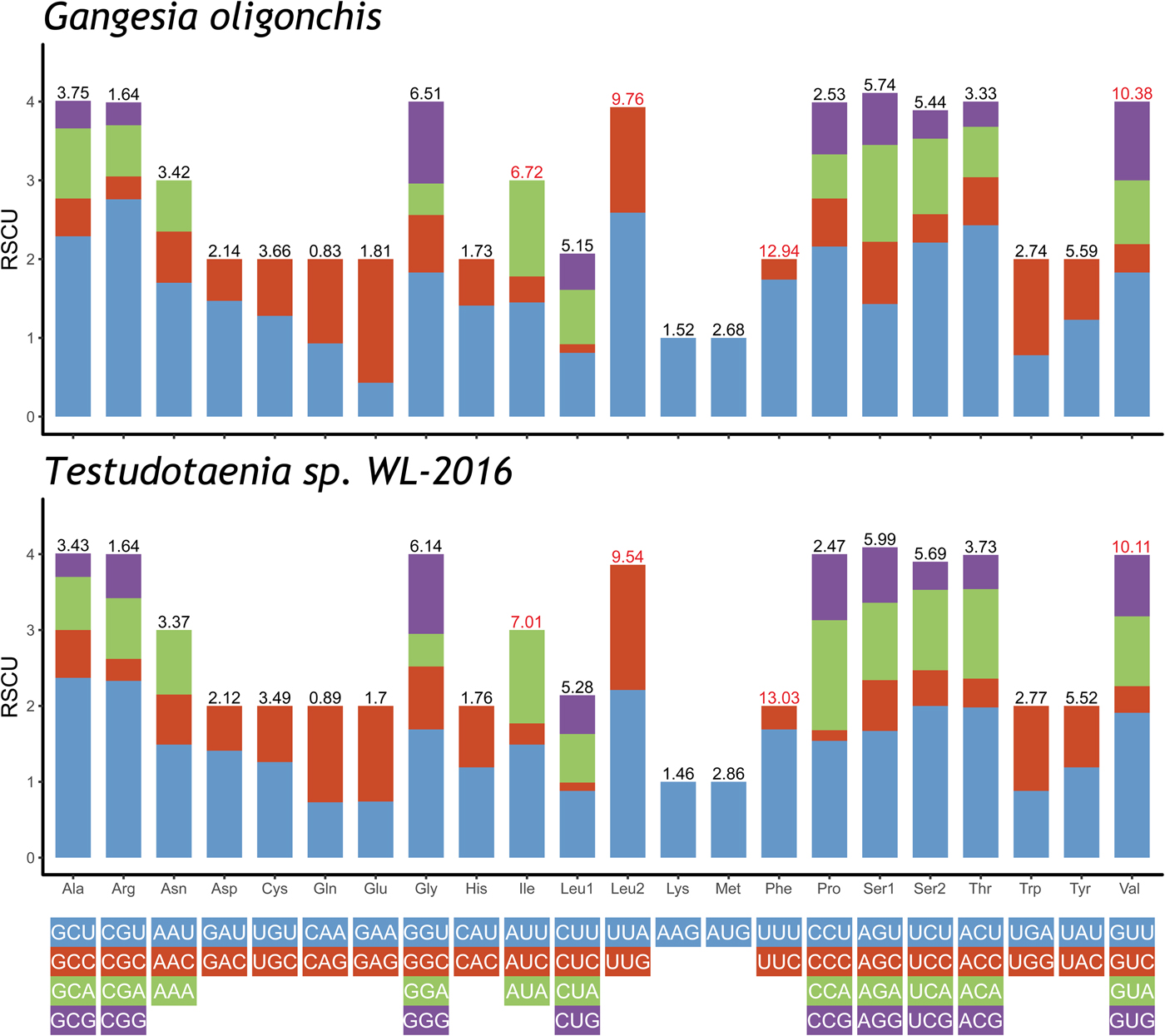
Fig. 2. Relative synonymous codon usage (RSCU) of the complete mitochondrial genome of Gangesia oligonchis and Testudotaenia sp. WL-2016. Codon families are labelled on the x-axis. Values on the top of the bars refer to amino acid usage.
Transfer and ribosomal RNA genes
All 22 tRNAs were found in the mitogenome of G. oligonchis; these ranged in length from 59 bp (trnS1) to 67 bp (trnN, trnM and trnG) (table 2). In terms of secondary structure, most of the tRNA sequences could be folded into the conventional cloverleaf shape; the exceptions were trnS1 and trnR, which lacked the dihydrouridine arms and loops. Standard anticodons were found in all tRNAs, except for trnR, which exhibited a transition from U to A. The genes rrnL and rrnS were 975 bp and 732 bp in length, with 66.7% and 67.2% A + T content, respectively (table 3). They were separated by trnC. The mitogenomic gene arrangement of trnT–rrnL–trnC–rrnS is shared by all cestodes characterized so far (fig. 3).

Fig. 3. The phylogenetic relationships of the five orders in Cestoda inferred from concatenated 36 genes representing almost complete mitogenomic datasets (36 genes: 12 PCGs, 2 rRNAs and 22 tRNAs), using two Trematoda species as outgroup. Scale bar represents the estimated number of substitutions per site. Bootstrap (BP)/posterior probability (BPP) support values of ML/BI analysis are shown above the nodes, only BP <100 and BPP <1 were displayed. Mitogenomic gene orders of the selected cestode species (corresponding to tip labels in the tree) were listed on the right of the tree. The order was reoriented to cox1.
Non-coding regions
A total of 22 short intergenic regions (1 to 12 bp in length) were interspersed within the mitogenome of G. oligonchis (table 2). These included two mNCRs consisting of an sNCR located between trnY and trnS2 and a lNCR located between Nad5 and trnG. These non-coding regions were 216 and 419 bp in size, respectively; they had a much higher A + T content at 78.7% and 84.3%, respectively (table 3). The sNCR and lNCR of Testudotaenia sp. WL-2016 (108 bp and 265 bp in size, respectively) were much smaller than those of G. oligonchis. Highly repetitive regions (HRRs) in lNCR were detected in both G. oligonchis and Testudotaenia sp. WL-2016, with 11 and six repeat units, respectively (fig. 4). Although the predicted stem (2 bp) of the Testudotaenia sp. WL-2016 repeat unit was extremely short, both consensus repeat units were capable of forming stem–loop structures (fig. 4). In addition, the sNCR of the two onchoproteocephalidean mitogenomes was also capable of forming a stem–loop structure (fig. 4).

Fig. 4. Illustration of highly repetitive regions in the large major non-coding region and the predicted secondary structure of the short non-coding region of Gangesia oligonchis and Testudotaenia sp. WL-2016 mitochondrial genome.
Phylogeny and gene order
Phylogenetic analyses of the concatenated 36 mitochondrial genes using BI and ML methods produced identical tree topologies in which G. oligonchis grouped as the sister taxon of Testudotaenia sp. WL-2016 with maximum support (BP = 100, BPP = 1). The ordinal topology is (Caryophyllidea, (Diphyllobothriidea, (Bothriocephalidea, (Onchoproteocephalidea, Cyclophyllidea)))). The mitogenomic gene arrangement of G. oligonchis was identical to that of Testudotaenia sp. WL-2016 and some cyclophyllideans, such as those of the families Hymenolepididae, Anoplocephalidae, Dipylidiidae and Paruterinidae (fig. 3).
Discussion
The topology of the trees resulting from phylogenetic analyses of the concatenated 36 mitochondrial genes was stable and in full agreement with those generated from studies based on complete mitogenomes (Li et al., Reference Li, Zhang, Boyce, Xi, Zou, Wu, Li and Wang2017) and large and small subunits of nuclear ribosomal RNA genes (28S rDNA and 18S rDNA) (Brabec et al., Reference Brabec, Kuchta and Scholz2006; Waeschenbach et al., Reference Waeschenbach, Webster and Littlewood2012; Kuchta & Scholz, Reference Kuchta and Scholz2017). However, this topology deviated from that recovered from the recent mitogenomic studies of Feng et al. (Reference Feng, Feng, Fang and Su2017) and Zhang et al. (Reference Zhang, Duan, Shi, Jiang, Zeng, Wang and Cui2017), in which a sister-group relationship of Diphyllobothriidea and Bothriocephalidea was recovered. These discrepancies may be caused by idiosyncrasies of different phylogenetic reconstruction software. To test this, another phylogenetic analysis was performed using the RaxML program (Silvestro & Michalak, Reference Silvestro and Michalak2011) (instead of MEGA) based on the same dataset and computational model as those of the studies by Feng et al. and Zhang et al. The resultant topology was identical to ours (see supplementary material fig. S1). Furthermore, a sister-group relationship of Bothriocephalidea and ‘acetabulate’ was also supported using a much denser taxon by 28S rDNA + 18S rDNA (Brabec et al., Reference Brabec, Kuchta and Scholz2006; Kuchta et al., Reference Kuchta, Scholz, Brabec and Bray2008) and partial mtDNA + 28S rDNA + 18S rDNA (Waeschenbach et al., Reference Waeschenbach, Webster and Littlewood2012; Kuchta & Scholz, Reference Kuchta and Scholz2017).
The mitogenomic gene arrangement of G. oligonchis was identical to that of Testudotaenia sp. WL-2016, which belonged to gene arrangement category IV (onchoproteocephalideans and some cyclophyllideans) as summarized by Li et al. (Reference Li, Zhang, Boyce, Xi, Zou, Wu, Li and Wang2017) (fig. 3). These authors concluded that all rearrangement events in cestode mitogenomes were observed in the rearrangement hot spot–P1 (i.e. gene block between rrnS and trnR), a region that often harbours an NCR (non-coding region). The TDRL (tandem–duplication–random–loss) event contributed to the increased rate of rearrangement for genes adjacent to the origin replication because both strand slippage and imprecise termination were more likely to include the genes surrounding the origin of replication in the rearrangement hot spot (Cameron, Reference Cameron2014). Despite the rearrangement hot spot in Cestoda, the mitogenomic gene arrangement was too conserved to reflect the inter-relationships within the order Onchoproteocephalidea.
Although mitogenomes are currently available for only two species of the order Onchoproteocephalidea, the low sequence identity (71.1%) between the mitogenomes of G. oligonchis and Testudotaenia sp. WL-2016 may provide some phylogenetic information. Within the family Diphyllobothriidae (Diphyllobothriidea), sequence identity of the mitogenome ranged from 85% to 87% between Ligula spp. and Dibothriocephalus spp. (Li et al., Reference Li, Fu, Zhang, Boyce, Xi, Zou, Li, Wu and Wang2018), which was much higher than that between G. oligonchis and Testudotaenia sp. WL-2016. Gangesia from catfishes, mostly in Indomalaya and Palearctic, is the early diverging group, while Testudotaenia from soft-shelled turtles in North America is the derived group (de Chambrier et al., Reference de Chambrier, Waeschenbach, Fisseha, Scholz and Mariaux2015). However, families and subfamilies within the Onchoproteocephalidea need to be determined based on mitogenomes of more taxa.
Conclusions
The complete mitogenome of the tapeworm Gangesia oligonchis from the bullhead catfish Tachysurus fulvidraco was sequenced and characterized. The mitogenomic gene arrangement was found to be conserved across the two members of the order Onchoproteocephalidea for which such data are available. While low nucleotide identity was found between the two onchoproteocephalideans, mitogenomes of more extensive taxa are expected to be sequenced to effectively explore the inter-relationships among the Onchoproteocephalidea.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S0022149X19000452
Author ORCIDs
W.X. Li, 0000-0001-7016-4535
Acknowledgements
The authors would like to thank Dr Jakovlić I. for his help in English language editing.
Financial support
This work was funded by the National Natural Science Foundation of China (31872604) and the earmarked fund for China Agriculture Research System (CARS-45-15).
Conflicts of interest
None.
Ethical standards
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional guides on the care and use of laboratory animals.









