


There are substantial associations between negative life events carrying long-term psychological threat and the onset of depressive disorder in adults (Reference Brown and HarrisBrown & Harris, 1978; Reference KesslerKessler, 1997). The same may apply in childhood and adolescence (Reference Goodyer and GoodyerGoodyer, 1995). However, there is huge individual variation in response. Although probably a majority of depressive disorders are associated with severely negative life events or experiences, only about one in five occurrences of severely negative life events leads to depression (Reference Brown, Bifulco and HarrisBrown et al, 1987). A key question concerns the origins of this individual variation in susceptibility to life events. Using an ingenious, but indirect, means of inferring genetic risk, Kendler et al (Reference Kendler, Kessler and Walters1995) showed that, in adults, negative life events were most likely to lead to onset of major depressive disorder in individuals inferred to have a genetic liability to depression. We set out to undertake a more direct test of the hypothesis that genetic factors moderate susceptibility to the environmentally mediated risks associated with negative life events.
Nature—nurture interplay
The starting point has to be the well-established finding that genetic factors have a role in individual differences in environmental risk exposure (Reference PlominPlomin, 1995; Reference Kendler and Karkowski-ShumanKendler & Karkowski-Shuman, 1997; Reference Rutter, Dunn and PlominRutter et al, 1997) and that the genetic liability to depression overlaps with the genetic liability to experience stressful life events (Reference Kendler and Karkowski-ShumanKendler & Karkowski-Shuman, 1997; Reference Silberg, Pickles and RutterSilberg et al, 1999). What this means is that, through their behaviour, people to some extent shape and select their environments.
This has been as evident in longitudinal studies as in genetic investigations. Thus, Champion et al (Reference Champion, Goodall and Rutter1995) found that children who showed emotional or behavioural disturbance on a teachers' questionnaire at age 10 years were more than twice as likely as those without disturbance to experience severely stressful events and experiences some 18 years later. The finding is entirely consistent with what is known about stressful life events and experiences (Reference RutterRutter, 2001). The psychopathological risks are greatest in the case of happenings that threaten important social relationships through loss, humiliation, entrapment, hostility or conflict. Because social relationships are dyadic and bidirectional, it follows that the ways in which one person behaves are likely to shape other people's responses and, hence, to increase or decrease the likelihood that the person will experience stressful life events.
This led to the crucial differentiation made by Brown & Harris (Reference Brown and Harris1978) between ‘independent’ and ‘dependent’ life events. It was appreciated from the outset that experiences people brought about through their own behaviour could still provide a psychopathological risk (see Reference Rutter, Silberg, Simonoff, Plomin and McLearnRutter et al, 1993). Nevertheless, in testing the hypothesis that stressful life events had a truly causal impact on the development of psychopathology, it was methodologically helpful to focus on those that, by their nature, were unlikely to have been brought about through the person's own behaviour.
This methodological innovation, together with others, was vitally important in establishing the case that life events were likely to play a causal role in the onset of depressive disorders (Reference Rutter and HarrisRutter, 2000). On their own, however, case—control designs are inadequate for the study of nature—nurture interplay in the susceptibility to environmental hazards. That is because, necessarily, the difference between depression groups and control groups in life events will be accompanied by parallel differences in both genetic liability and earlier exposure to other environmental risks (Reference Robins, Robertson and DohrenwendRobins & Robertson, 1998). Genetic designs are crucial in order to put the environmental mediation hypothesis under rigorous test. The solution lies in a focus on the association between life events and psychopathology within monozygotic twin pairs, who do not differ in genetic liability; in treating life events as a phenotype in bivariate analyses that use both cross-twin and cross-trait associations; and in studying life events that involve no genetic mediation (Reference Kendler, Karkowski and PrescottKendler et al, 1999).
In twin designs, it is not possible to identify gene—environment interactions (G × E) in an unambiguous manner if there are also gene—environment correlations (rGE). Consequently, if nature—nurture interplay is to be analysed in its various facets, it will be necessary to adopt different strategies for the investigation ofG × E (genetically influenced sensitivity to the environment) and rGE (an association between genetic and environmental risks).
In order to study G × E, we examined life events with a demonstrated empirical association with depression/anxiety within our own data-set (i.e. those for which there was a prima facie case for possible environmental mediation of risk), focusing on samples in which there was also empirical evidence of genetic liability for depression/anxiety (this meant restriction to adolescent girls). The next step was to subdivide these events into those that, by their nature, seemed dependent and those that seemed independent. The rationale was that rGE was likely to be operative in the former and absent in the latter, although that assumption had to be tested. The former provided the opportunity to study the role of rGE in the liability to depression/anxiety. The findings showed that dependent life events were significantly associated with depression within monozygotic twin pairs (Reference Silberg, Pickles and RutterSilberg et al, 1999; Reference Rutter and HarrisRutter, 2000); therefore, despite being brought about by the person's own behaviour, the experience exerted true environmentally mediated risk for depression from life events.
The next stage was to focus on independent life events (for whichrGE was unlikely) in order to test for G × E. The postulate was that the depression/anxiety would be more likely to be associated with negative life events in individuals with a genetic liability. The research strategy involved four interconnected steps. First, it was necessary to establish that there was a significant effect of life events on depression/anxiety and that there was no appreciable genetic effect on such life events. Second, we tested for a significant G × E interaction such that the genetic effect was greater in the presence of life events, indicating that an individual's inherited liability to symptoms of depression (i.e. sensitivity) is affected by exposure to certain environments. Although it sounds paradoxical, that implies genetic moderation of sensitivity to life events. That is because, according to standard practice, all effects of G × E are included in the genetic term. Accordingly, the third step was to split the genetic term into the effects of baseline genes and the effect of G × E. If the genetic moderation of environmental risk mediation was correct, the whole of the difference in genetic effect according to the presence or absence of life events should be accounted for by G × E. The fourth step involved testing whether the effect of life events was greater in the presence of parental emotional disorder. Clearly, parental disorder will index more than just genetic risk, but if the genetic moderation hypothesis was correct it should follow that the effect of life events would be greater in the presence of parental emotional disorder than in its absence.
METHOD
The analysis was based upon data collected on girls aged 14 years or more from the Virginia Twin Study of Adolescent Behavioral Development (VTSABD). Details concerning the design, ascertainment, assessment protocol and participation rates for this study are provided elsewhere (Reference Hewitt, Silberg and RutterHewitt et al, 1997; Reference Simonoff, Pickles and MeyerSimonoff et al, 1997). Because our earlier findings had shown that genetic effects on depression were greater in pubertal adolescents than in children and greater in females than in males, we focused on girls who were likely to be well advanced in the transition through puberty, i.e. at least 14 years old (Reference Silberg, Pickles and RutterSilberg et al, 1999). Our analyses had also shown that this was an age period when life events were likely to be having an effect on depression and anxiety. There were 184 same-gender female pairs, with two waves of data collection some 15 months apart.
Assessment of depression and anxiety
Symptoms of depression and generalised anxiety disorder were assessed according to their presence in the 3 months immediately preceding interview, using the child's responses to the Child and Adolescent Psychiatric Assessment (CAPA; Reference Angold, Prendergast and CoxAngold et al, 1995). This is an investigator-based psychiatric interview that provides information for diagnosing the major forms of childhood psychopathology according to DSM—IV criteria (American Psychiatric Association, 1994). Each symptom diagnostic of either a major depressive episode or generalised anxiety disorder was coded as if the symptom were present in at least two areas of activity in the child's life and at least somewhat uncontrollable. Otherwise it was considered not to be present. The respective symptoms were then summed into a depression and anxiety sub-scale for subsequent data analysis.
Selection of life events
A life events scale was composed from the maternal ratings of events in the preceding year (Reference Johnson, McCutcheon, Sarason and SprelbergerJohnson & McCutcheon, 1990) that were significantly associated with depression, but were considered to be beyond the child's control (independent life events). Dependent life events (events potentially within the child's control) have been shown to be genetically correlated with risk to depression in this sample (Reference Silberg, Pickles and RutterSilberg et al, 1999). Theoretically, if particular life events are linked to a common set of genes that also influence risk to depression (a genotype × environment correlation), there could be increasing genetic variance for depression in the presence of those same events. Because the inclusion of events that are dependent on the child's genotype can potentially give rise to the same pattern of results as a gene—environment interaction, for clarity of interpretation only independent life events were included in theG × E interaction analysis.
Since a G × E interaction is considered to be operating only if there are increasing genetic differences as a function of differences in exposure to a true environmental risk factor, the twin correlations for the independent life events were also estimated in monozygotic (MZ) and dizygotic (DZ) post-pubertal females using SAS (SAS Institute, 1996).
Epidemiological analyses
The PROC MIXED procedure in SAS was used to select life events significantly related to symptoms of depression. The procedure takes account of the clustering of observations from twins of a pair and across the two waves of data as part of the fitted model. The PROC LOGISTIC procedure was then used to estimate the odds ratio of life events to symptoms of depression and anxiety using the top 10% of scores as a cut-off for generating a categorical index of the two traits.
Model fitting
Raw data collected from the female twin pairs from the first and second waves of data were analysed by maximum likelihood using Mx 1.47 (details available from M. N. upon request). The basic genetic model comprises additive genetic (A), common (C) and specific (E) environment components of variation. In the G × E interaction model (diagrams of the model are available from the author upon request), we examine whether the genetic factors contribute the same amount to the variance under different environmental conditions, in this case the measured level of independent life events. We use a multi-level extension of this method (Reference Neale, Cardon, Neale and CardonNeale & Cardon, 1992) by specifying a different model according to the level of stress assessed in each twin. The covariance between depression and anxiety is modelled as one factor that influences both traits, and a second factor that influences anxiety only. The genetic part of the model is subdivided into two components: a ‘baseline’ component and a component that varies according to the stress level. By comparing the fit of the model with the moderating effect of stress with that of a model without moderating effects, we can judge the statistical significance of the moderation (interaction) using a likelihood ratio test. The difference between —2(log likelihood) of the two models is distributed as a chisquare (χ2), and degrees of freedom (d.f.) are calculated by subtracting the number of estimated parameters of a reduced model from those estimated in the fuller model. To avoid a false-positive finding when testing for a moderating effect of the environment, the main effect of life events is regressed out of both the depression and anxiety sub-scales just prior to model fitting.
A critical test of the G × E interaction model is that the data cannot be explained by stress-related differences in the specific environment. This could arise from heteroscedasticity, or increasing error variance in groups of individuals with higher mean levels of stress. A model that allowed the unique environmental parameter to differ in the various exposed groups was compared with the fit of the G × E interaction model that allowed for differences in genetic variance as a function of environmental exposure. To assess the moderating effect of life events on the shared environmental variance, a model that allowed this parameter to vary across the different life event groups was also fitted to the data on depression and anxiety and similarly evaluated against theG × E interaction model.
As another test of a G × E interaction, maternal and paternal history of generalised anxiety disorder or major depressive disorder (assessed on the basis of systematic interview data) was used as an index of familial risk for depression and anxiety in the child. Although parental psychopathology could represent a genetic or shared environmental liability (or more probably a combination of the two), if a G ×E interaction were operating, we would expect an increased risk for depression or anxiety as a function of the presence of life events and psychiatric disorder in one or both parents. The PROC MIXED procedure was used to test for mean differences in depression and anxiety as a function of life events in those with and without a history of parental anxiety/depression.
RESULTS
Three past-year events reported by the girls' mothers were significantly related to child-reported depression. These were:
-
(a) a new stepbrother or stepsister;
-
(b) brother or sister (or stepbrother/stepsister) leaving home;
-
(c) the father losing his job.
Taken together, these three events had a correlation of 0.11 with depressive symptoms and 0.06 with anxiety symptoms, comparable to odds ratios of 1.5 and 1.3, using a dichotomised measure of the two traits. This phenotypic association indicated that life events had an effect on emotional difficulties in adolescent girls, statistically significant (P=0.001) in the case of depression and just short of significance for anxiety (P=0.07). There was a need to accompany this demonstration with a test of whether it was likely that genetic mediation could be involved. All three events were of a kind that should be shared across twins within the same pair and hence very high within-pair correlations were to be expected. The question was whether, despite sharing, there was a difference between MZ and DZ pairs. The findings showed that there was not. The average correlation for the life events sub-scale across the two waves was 0.91 for MZ twins and 0.87 for DZ twins, evidence that these life events are not dependent upon the genotype of the child, but were truly environmental in nature.
Having shown that the life events had a significant effect on depression/anxiety and that this represented environmental mediation, the next step was to test for G × E interaction. A model that allowed for increasing genetic variance as a function of environmental exposure fitted relatively better than a model that allowed for increasing unique environmental variance or shared environmental variance as a function of number of life events (further details available from the author upon request). The model fitting results shown in Table 1 indicate that eliminating the interaction term from the full G × E interaction model (Model II) resulted in a significant deterioration in fit for both depression (Model I) and anxiety (Model III). These results also showed that exposure to life events increases the genetic association or genetic covariance between the two traits (Model IV).
dTable 1 Results of fitting genotype × environment (G ×E) interaction models to depression and anxiety

| Model | -2LL | d.f. | χ 2 difference | P | |
|---|---|---|---|---|---|
| I | Full G× E interaction model | 4440.029 | 2059 | ||
| II | Drop G× E interaction for depression | 4456.278 | 2061 | 16.25 | 0.000 |
| III | Drop G× E interaction for anxiety | 4450.158 | 2061 | 10.13 | 0.006 |
| IV | Drop G× E interaction for covariance between depression and anxiety | 4444.476 | 2060 | 4.45 | 0.035 |
The better fit of the G × E interaction model indicates that there are genetically mediated differences in adolescent girls' sensitivity to the effects of the environment. The proportions of overall variance in depression and anxiety due to genetic effects for each level of life event from this model are depicted in Fig. 1. For depression, the genetic variance rose from 0.27 in the absence of life events, to 0.33 for one life event and to 0.39 for two life events. The comparable estimates for anxiety were 0.19, 0.34 and 0.44 respectively. Although there was a trend for greater moderation of genetic variance for anxiety in the presence of one or more life events, it is not significantly different from the increase in the environmentally influenced change in genetic variance observed for depression.
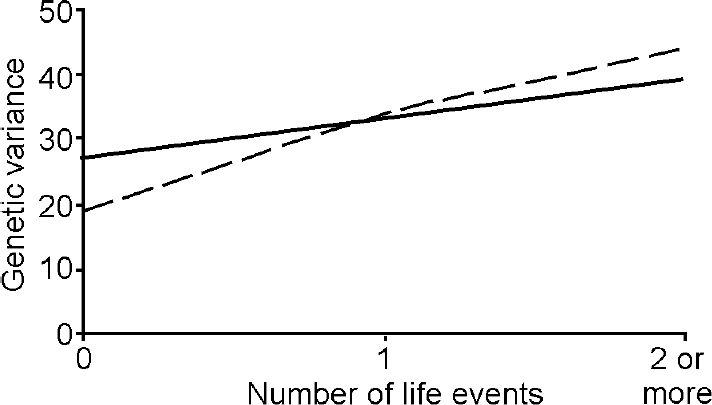
Fig. 1 Change in genetic variance for depression (solid line) and anxiety (dashed line) as a function of number of past-year life events.
The third step in the analysis involved determination of whether the difference in the genetic term according to the presence of life events was true owing to G × E. The components of variance for depression and anxiety attributable to baseline genes and G ×E interaction under different levels of events are shown in Figs 2 and 3, which demonstrate that all of the change in genetic variance for depression and anxiety in the presence of one, two or more life events was due to the effect of the interaction between genes and environment.
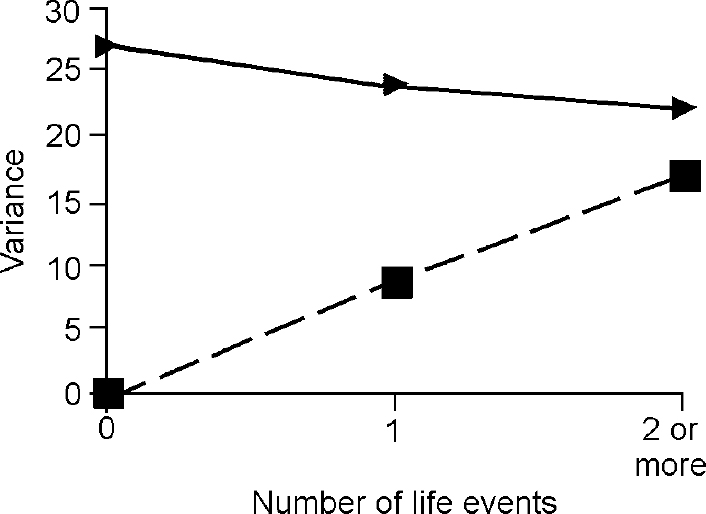
Fig. 2 Effect of life events on variance in depressive symptoms accounted for by ‘baseline’ genetic effects (triangles) and by gene—environment interaction (squares)
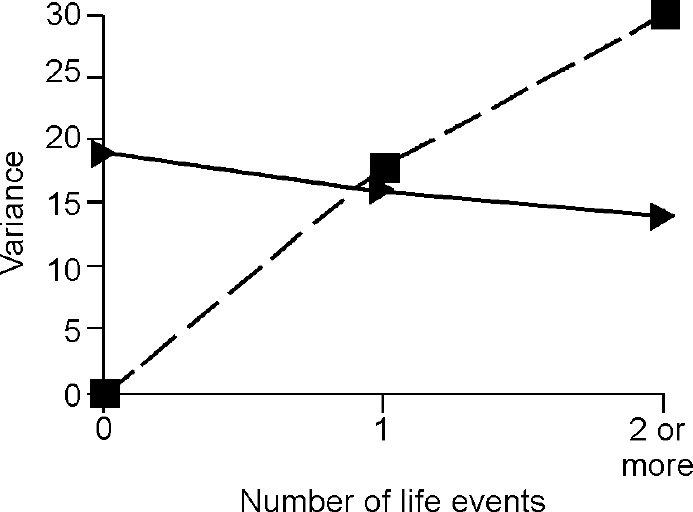
Fig. 3 Effect of life events on variance in anxiety symptoms accounted for by ‘baseline’ genetic effects (triangles) and by gene—environment interaction (squares).
The fourth and final step involved testing of the effects of life events in the presence and absence of parental emotional disorder.Table 2 presents the average CAPA depression and anxiety scores (standardised to a mean of 10 with a standard deviation of 3) contingent on exposure to life events and parental psychopathology, an index of familial risk. The first finding was that there was no significant — or even appreciable — effect of life events when there was no emotional disorder in either parent (9.57 v. 9.56,P=0.98; 9.52 v. 9.60, P=0.85). The strong implication is that for life events to have any causal effect on depression/anxiety in adolescent girls there must be either a genetic predisposition or an increased vulnerability resulting from the environmental effects of parental emotional disorder. Without these, there was no demonstrable psychopathological risk associated with the life events studied. The second finding was a significant effect of life events on depression (10.05 v. 10.98; P=0.01) when these occurred against the background of parental emotional disorder (the trend for anxiety was similar but fell well short of significance, P=0.15). This suggests that, given genetic or environmental vulnerability, life events do have an important environmental role in the causation of depressive symptomatology. The third finding was that parental emotional disorder had an effect on depression/anxiety both in the absence of life events (9.57 v. 10.05,P=0.04 for depression; 9.52 v. 10.16, P=0.007 for anxiety) and in its presence (9.56 v. 10.98, P=0.02 for depression; 9.60 v. 10.64, P=0.04 for anxiety). The implication is that part of the genetic mediation involves G ×E but much does not (at least in so far as the life events indexed environmental risk). The pattern of results for depression is consistent with a G × E interaction — the girls with the highest depression scores are those who experienced a life event in the past year and who had a mother or father (or both) with a history of depression or anxiety.
Table 2 Level of depressive and anxiety symptoms in adolescent girls according to parental emotional disorder and presence of one (or more) life events
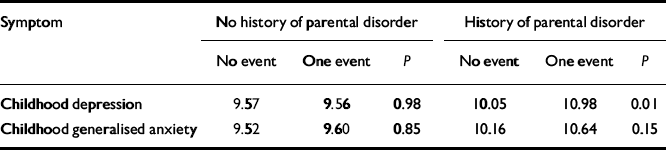
| Symptom | No history of parental disorder | History of parental disorder | ||||
|---|---|---|---|---|---|---|
| No event | One event | P | No event | One event | P | |
| Childhood depression | 9.57 | 9.56 | 0.98 | 10.05 | 10.98 | 0.01 |
| Childhood generalised anxiety | 9.52 | 9.60 | 0.85 | 10.16 | 10.64 | 0.15 |
DISCUSSION
Despite the remarkable degree of individual variation in response to psychosocial experiences, surprisingly little is known about the causal mechanisms underlying this variation. This applies as much to the effects of negative life events on liability to depression, as to the other experiences. In order to determine why people vary in their susceptibility to environmental stressors, it is crucial to start with a research strategy that can provide a rigorous test of environmental mediation of psychopathological risk. Until such mediation is established, it is not possible to be sure that susceptibility to psychosocial risks is truly being examined. That has posed a substantial limitation in view of the evidence that there are important gene—environment correlations (Reference PlominPlomin, 1995) and, hence, that the risks associated with negative events and experiences could be genetically mediated in part. To rule out genetic mediation, it is desirable to use genetically sensitive designs.
In studying the effects of negative life events on depressive and anxious symptomatology in adolescent girls, we used a twin design that allowed us to use two different tests of environmental mediation. First, focusing on child-specific events, we had shown previously that life events were significantly associated with depression even within MZ pairs, who shared the same genetic risks (Reference Silberg, Pickles and RutterSilberg et al, 1999; Reference Rutter and HarrisRutter, 2000). Second, by focusing on life events that impinged equally on both twins, we confirmed that there was no significant within-pair genetic effect on such life events, and that the significant effect of life events on depression/anxiety had therefore to be environmentally mediated.
In keeping with other studies, we found major individual variation in susceptibility to life events. Only a minority of the adolescent girls experiencing life events showed depression/anxiety. We tackled the hypothesis that part of the individual variation in susceptibility could be genetically influenced by testing for G × E interaction. Significant interaction was found. Genetic effects on depression/anxiety, and especially on their co-occurrence, were substantially and significantly greater in individuals who had experienced negative life events in the past year than in those without such experiences. We went on to show that this difference in genetic effect was due to G × E interaction, there being no difference in the effects of baseline genes.
Finally, we employed a quite different test of interaction by comparing the effects of life events on depression/anxiety in the presence and in the absence of parental emotional disorder. The findings showed no effect of life events in girls whose parents had not experienced emotional disorder, a result that is entirely consistent with the hypothesis that G ×E interaction accounts for much of the individual variation in susceptibility to life events. Equally, we found that the effects of parental emotional disorder were significantly greater in the presence of life events than in their absence, again pointing to the likely role of G ×E interaction. On the other hand, there was a significant effect of parental mental disorder in the absence of life events, indicating that life events did not constitute the only route of risk mediation.
In order to test for G × E interaction, it was necessary for methodological reasons to focus on a subset of life events that did not show a gene—environment correlation. Almost certainly that resulted in our studying life events with a relatively weak risk effect on depression/anxiety. In order to provide a better estimate of the overall environmentally mediated risk for depression/anxiety associated with life events, it will be necessary to use bivariate analyses, treating total life events exposure as a phenotype. That will constitute the next step in our overall strategy to study the effects of life events. However, that method will not allow any satisfactory investigation of G × E interaction (the focus of this study).
The findings do not, of course, mean that the whole of individual variation in susceptibility to environmental risks is due to G ×E interaction. It is highly likely that both previous environmental risk exposure and concurrent exposure to other more long-standing psychosocial adversities also play a role in individual variations in susceptibility. The investigation of that possibility will require the use of different designs. Also, greater leverage on the operation of environmental risks can be obtained by using the three waves of data collection (spanning some 3 years) available in VTSABD and by putting together a greater range of psychosocial risk factors.
Our study of the effects of life events had to rely on a questionnaire measure, which is inevitably cruder than the detailed investigator-based methods that have come to be accepted as the optimal approach. The consequence is that the true effect of life events on depression/anxiety may well be greater than that found here. On the other hand, our reliance on a questionnaire measure is unlikely to have biased our finding of a significantG × E interaction. We took particular care to use methods that avoided the possibilities of criterion contamination deriving from reliance on a single informant for both independent and dependent variables — a major limitation of most life events studies (Reference RutterRutter, 2001). Thus, we used parental reports for life events and child reports for depression/anxiety. A further problem in much life events research has been the possibility that the life events may have been caused by the person's behaviour, rather than the other way round. Both our selection of life events and our use of a twin design effectively ruled out that possibility. Our data were cross-sectional rather than longitudinal and, hence, we could not focus specifically on the role of life events in provoking the onset of disorder. However, that was not our goal. Rather, our focus was on the liability to depression/anxiety, for which the methods used were well suited. Nevertheless, longitudinal data, to which we will turn next, will help in sorting out causal processes because they will provide a better leverage on recurrent/chronic symptomatology and because they will allow us to examine the extent to which the psychopathological risks associated with life events derive from their accumulation over time rather than their occurrence during a specified period prior to the onset of a new disorder.
A further query concerns the nature of the phenotypic individual feature that indexes susceptibility to environmental stressors. Psychosocial researchers have tended to favour explanations in terms of cognitive sets (Reference Brown and HarrisBrown & Harris, 1990) whereas behavioural geneticists (Reference Kendler, Karkowski and PrescottKendleret al, 1999) and others (Reference AndrewsAndrews, 1996) have usually focused on the personality trait of neuroticism. These (and other) alternatives need to be put to the test, not just in relation to a general liability to depression/anxiety but specifically in relation to their possible role as influences on susceptibility to life stressors.
Meanwhile, the message from our findings is that genetic factors have a significant role in the individual response to environmental stressors. It is also evident that genetic effects on liability to depression/anxiety must be considered as operating through indirect, as well as direct, routes. That is, they operate in part because they influence both exposure to environmental risks (gene—environment correlations) and susceptibility to the psychopathological effects of risk environments (gene—environment interactions). Traditional forms of analysis attribute the whole of both these indirect effects to genes, but it is apparent that this is misleading. The psychopathological risks come about as a specific result of the conjunction, and joint operation, of genetic and environmental influences. The task of delineating the causal mechanisms underlying the interplay between nature and nurture constitutes a major research priority for the future. The well-known finding of increasing depression in adolescent girls may be due to increasing genetic differences in sensitivity or reactivity to the environment.
Clinical Implications and Limitations
CLINICAL IMPLICATIONS
-
• Negative life events have an environmentally mediated effect on depressive/anxious symptomatology.
-
• Individual differences in susceptibility to stressful life events are due, in part, to genetic influences.
-
• Psychosocial risks for depression/anxiety are most likely to be operative in individuals who are also at genetic risk.
LIMITATIONS
-
• Life events were measured by questionnaire rather than interview.
-
• For unavoidable methodological reasons it was necessary to focus on a subset of independent life events.
-
• Analyses were based on cross-sectional, rather than longitudinal, associations.






eLetters
No eLetters have been published for this article.