


Diphtheria, the classical disease caused by Corynebacterium diphtheriae, is an acute communicable infection of the upper respiratory tract that can be fatal [Reference Hadfield1]. Although the global incidence of this disease has reduced dramatically as a result of increased vaccine coverage, infections by the different biotypes of C. diphtheriae continue to be reported in many countries [Reference Mattos-Guaraldi2, Reference Wagner3]. Moreover, the emergence in recent years of non-toxigenic strains of this bacterium as causative agents of severe invasive infections in vaccinated and non-vaccinated individuals has raised major concerns [Reference Wagner3, Reference Romney4].
Most of the reports in the literature regarding invasive infections caused by C. diphtheriae are related to infective endocarditis [Reference Mishra5]. Mortality rates are estimated to be high (43% in the cases reviewed by Mishra et al. [Reference Mishra5]), and predisposing factors such as incompetent cardiac valves and intravenous drug use apparently contribute to most cases [Reference Mishra5]. Nonetheless, endocarditis caused by C. diphtheriae has also been reported in individuals without recognized risk factors [Reference Hirata6].
Microbiological studies indicate that C. diphtheriae biotypes gravis and mitis are attributed to invasive diseases at higher rates (97·5% of the cases reviewed by Mishra et al. [Reference Mishra5]), while infections by intermedius and belfanti biotypes are much less common [Reference Mishra5]. Regardless of the biotype, most infections for which information is available were apparently caused by non-toxigenic strains of C. diphtheriae [Reference Mishra5]; molecular-typing methods were used to identify specific clones of this bacterium as the causative agents of such invasive infections in only a few examples [Reference Romney4, Reference Gubler7].
In the present work, we investigated the genetic relationships between invasive C. diphtheriae strains isolated in the urban area of Rio de Janeiro, Brazil, during a 10-year period (from 1993 to 2003).
Clinical and microbiological data on the C. diphtheriae invasive strains studied are shown in Table 1. All the strains (n=6) rendered negative results in PCR reactions with previously published primers [Reference Pimenta8] which target the tox gene (Table 1); amplification of the dtxR gene was used as a species-specific marker, as described previously [Reference Pimenta8]. Another characteristic of some of these C. diphtheriae invasive isolates (n=4) is the atypical sucrose-fermenting ability (Table 1) [Reference Hirata9]. The sucrose-positive phenotype has also been observed in a strain causing classical diphtheria in Brazil (strain TR241) [Reference Hirata9]; therefore, this strain was included in the study along with a sucrose-negative strain (VA01) which also caused typical disease and was isolated in the same geographical region (Table 1). The sucrose-negative C. diphtheriae strains ATCC 27012 and CDC E8392 were used as controls.
Table 1. Clinical and microbiological information on the strains of Corynebacterium diphtheriae used in the study and results of the MLST analysis
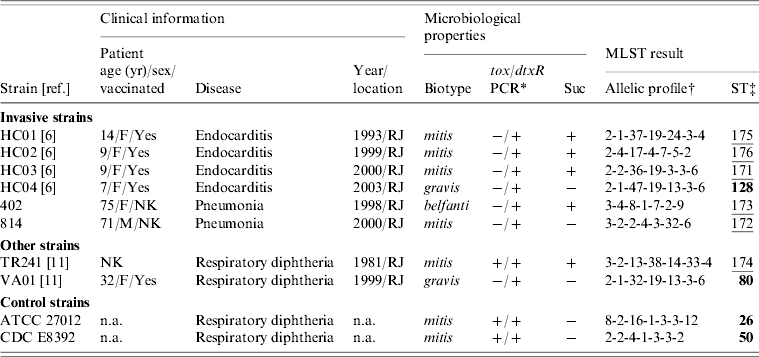
Suc, Sucrose fermentation; NK, not known; n.a., not applicable (additional information available from the American Type Culture Collection, USA, or the Centers for Disease Control and Prevention, USA).
* PCR reactions targeting the tox gene of C. diphtheriae were performed according to Pimenta et al. [Reference Pimenta8].
† Allelic order: atpA, dnaE, dnaK, fusA, leuA, odhA, rpoB.
‡ Novel sequence types (ST) are underlined. Numbers in bold indicate a match with previously identified STs.
We used a recently introduced multilocus sequence typing (MLST) scheme [Reference Bolt10] for molecular characterization of C. diphtheriae strains. This method types strains by indexing nucleotide variation within seven housekeeping genes: atpA, dnaE, dnaK, fusA, leuA, odhA and rpoB. After amplification, the respective genes' sequencing reactions were performed using a nested approach, according to a standardized protocol [Reference Bolt10]. Allelic profiles and sequence type (ST) designations for each studied strain were obtained by depositing the generated DNA sequences in the PubMLST database (http://pubmlst.org/cdiphtheriae/).
Of the six invasive C. diphtheriae strains studied five were new STs (Table 1). Only the sucrose-negative strain HC04, isolated from a case of endocarditis in 2003, generated a MLST profile that had been previously deposited in the database, ST128. Interestingly, this was a single locus variant (SLV) of ST80 at the dnaK locus, indicating that these two STs are clonally related; this latter ST was assigned in this study to a sucrose-negative strain (VA01) previously recovered from a case of typical diphtheria that occurred in a patient that had regular contact with European travellers during the last 5 days before onset of symptoms [Reference Mattos-Guaraldi11]. The fact that both strains (HC04 and VA01) were assigned to already known STs means that they are part of a clonal complex that comprises strains isolated in different countries including France (ST128) and Canada (ST80) (Fig. 1). The C. diphtheriae control strains used in this study, both from North America, were also assigned to known MLST profiles (Table 1).
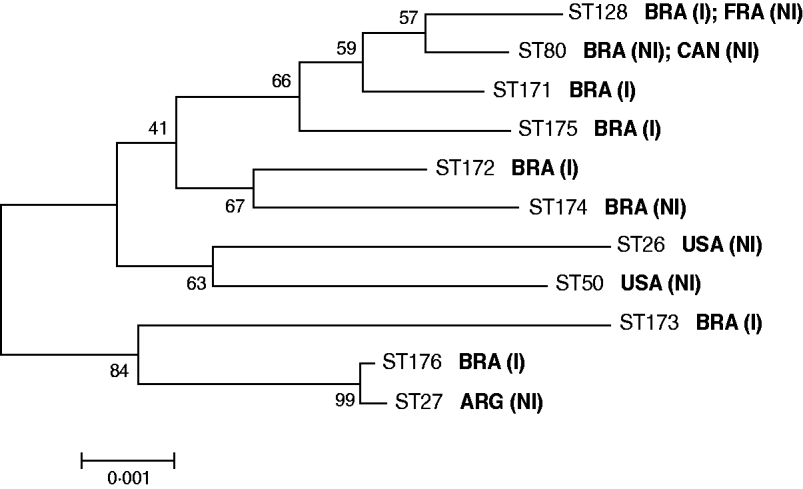
Fig. 1. Neighbour-joining phylogenetic tree showing the genetic relationships between the sequence types (STs) of C. diphtheriae studied. Concatenated MLST sequences were used for this analysis. The countries of isolation of the different C. diphtheriae strains are indicated. I, Isolate from invasive disease; NI, isolate from non-invasive disease (typical diphtheria).
In order to study further the clonal relationships between invasive C. diphtheriae strains, we performed analyses using the eBURST V3 program (http://eburst.mlst.net/). The analyses also included 12 previously identified STs [Reference Bolt10], which were representative of different C. diphtheriae biotypes circulating during different years in The Americas, Europe, Africa and Asia (data not shown). Two STs were considered as being part of a clonal complex if they shared at least six of the seven MLST alleles studied, as described previously [Reference Bolt10].
eBURST analysis demonstrated that invasive C. diphtheriae strains isolated in the metropolitan area of Rio de Janeiro, Brazil are part of distinct clonal complexes. This result is different from results reported in other countries, where invasive infections were apparently related to specific clones [Reference Romney4, Reference Gubler7]. In addition, the analysis was also not indicative of clonal relationships between all the sucrose-positive isolates of C. diphtheriae. This may be due to some recombinogenic potential of the strains, as suggested by the index of association (IA) between alleles at the different MLST loci, calculated according to Bolt et al. [Reference Bolt10]: IA sucrose-positive strains=0·0283; IA all invasive strains=0·1508.
Of note, ST176 (strain HC02; biotype mitis isolated in 1999) formed a clonal complex with ST27, which was previously assigned to a toxigenic biotype mitis strain isolated in Argentina in 1995 causing classical diphtheria (Fig. 1). This result may be indicative of a specific C. diphtheriae biotype mitis clonal complex with circulation associated with South America, as has also been suggested in previous studies [Reference Mattos-Guaraldi2], although this warrants further investigation. Most importantly, MLST results obtained for strains HC02 (ST176) and HC04 (ST128) demonstrate that C. diphtheriae strains causing invasive infections in Brazil can be normally found in clonal complexes with strains related to non-invasive disease (Fig. 1). Genetic and microbiological factors leading to the invasive phenotype also need further study [Reference Romney4, Reference Hirata6].
Hirata and co-workers evaluated microbiological aspects that might be related to endocarditis caused by some of the C. diphtheriae strains used in the present study [Reference Hirata6]. They found a characteristic adherence phenotype of the invasive strains, which apparently correlated with specific protein profiles in SDS–PAGE gels. Moreover, this same group showed that the capacity to bind to Fbn and convert Fbn to fibrin may play a role in pseudomembrane formation and act as a virulence determinant of both non-toxigenic and toxigenic strains [Reference Sabbadini12]. Further studies are underway to evaluate the contribution of these factors in development of invasive infections. A preliminary analysis of genetic diversity of five Brazilian C. diphtheriae invasive strains was performed using random amplification of polymorphic DNA (RAPD) [Reference Hirata6], which has been suggested recently to be as discriminatory as ribotyping, previously regarded as the ‘gold standard’ method for analysis of C. diphtheriae, until the emergence of MLST. Our MLST results were in accordance with the previous RAPD results [Reference Hirata6], in which four of the invasive strains studied had already been proposed to be part of different clonal groups. Besides extending this analysis to novel strains, the intrinsic advantages of MLST over other molecular-typing methods allowed for a better understanding of the genetic relationships between invasive and non-invasive strains of C. diphtheriae isolated in Brazil.
ACKNOWLEDGEMENTS
This work was funded in part by CNPq, CAPES and FAPERJ in Brazil, the Medical Research Fund (to C.G.D.) in the UK, and the Institut Pasteur in France.
DECLARATION OF INTEREST
None.


