



Case 1
A 3-year-old male patient was referred when he was 3 months old because of a cardiac murmur heard on physical examination. The patient’s fetal echocardiographic revealed biventricular hypertrophy, but he did not return for postnatal follow-up. There was no history of heart disease in the family. An X-ray of the chest revealed mild cardiomegaly. Electrocardiography demonstrated no abnormalities other than a high precordial voltage, and a 24-hour rhythm Holter examination showed no arrhythmia. An echocardiogram revealed significant biventricular hypertrophy and a left ventricular outflow tract obstruction with preserved systolic function (Fig. 1). Propranolol and captopril treatment was initiated since he had moderate mitral valve regurgitation. Serum creatinine kinase and liver transaminases were slightly elevated. Other metabolic tests, such as acid maltase activity, were normal. Danon disease was identified after the discovery of a homozygous deletion in the LAMP-2 gene. Neurologic and ophthalmologic examinations were found to be normal. His electrocardiography revealed a Wolff-Parkinson-White pattern, which was absent before at the age of 23 months. The asymptomatic patient is being followed up with medications.
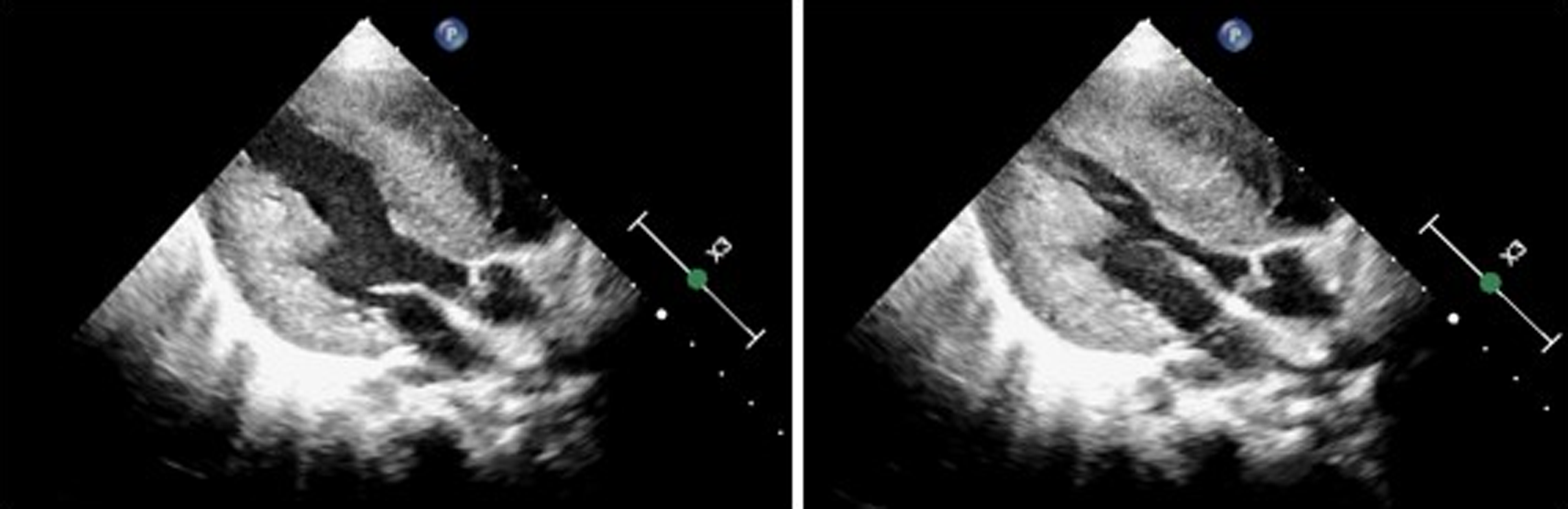
Figure 1. Echocardiography shows marked concentric hypertrophy and left ventricular outflow tract obstruction.
Case 2
A 16-year-old female patient was admitted with a complaint of fatigue. Except for an anxiety disorder, her background was unremarkable. Her father’s male cousins had a history of sudden death unexpectedly. Electrocardiogram revealed a short PR interval, a delta wave, ST depression, and left ventricular hypertrophy. Echocardiographic study demonstrated concentric left ventricular hypertrophy, diastolic dysfunction, and mild mitral regurgitation, with preserved systolic functions. Metoprolol and enalapril treatment was initiated after non-sustained ventricular tachycardia attacks were detected in 24-hour Holter monitoring (Fig. 2). In routine laboratory tests, lactate dehydrogenase and liver transaminases were slightly elevated. Acid maltase activity and metabolic tests were both normal. The fasciculoventricular accessory pathway was detected in the electrophysiological study. Danon disease was diagnosed after genetic test revealed an exon 4–5–6 deletion in the LAMP-2 gene. Electromyography revealed no evidence of a myopathy pattern. Other than myopia and astigmatism, no other findings were discovered during the ophthalmological examination. Amiodarone was added to the treatment of a patient who had syncope and continued to have non-sustained ventricular tachycardia during the first year of follow-up. The patient’s family refused to have a cardioverter defibrillator implanted.
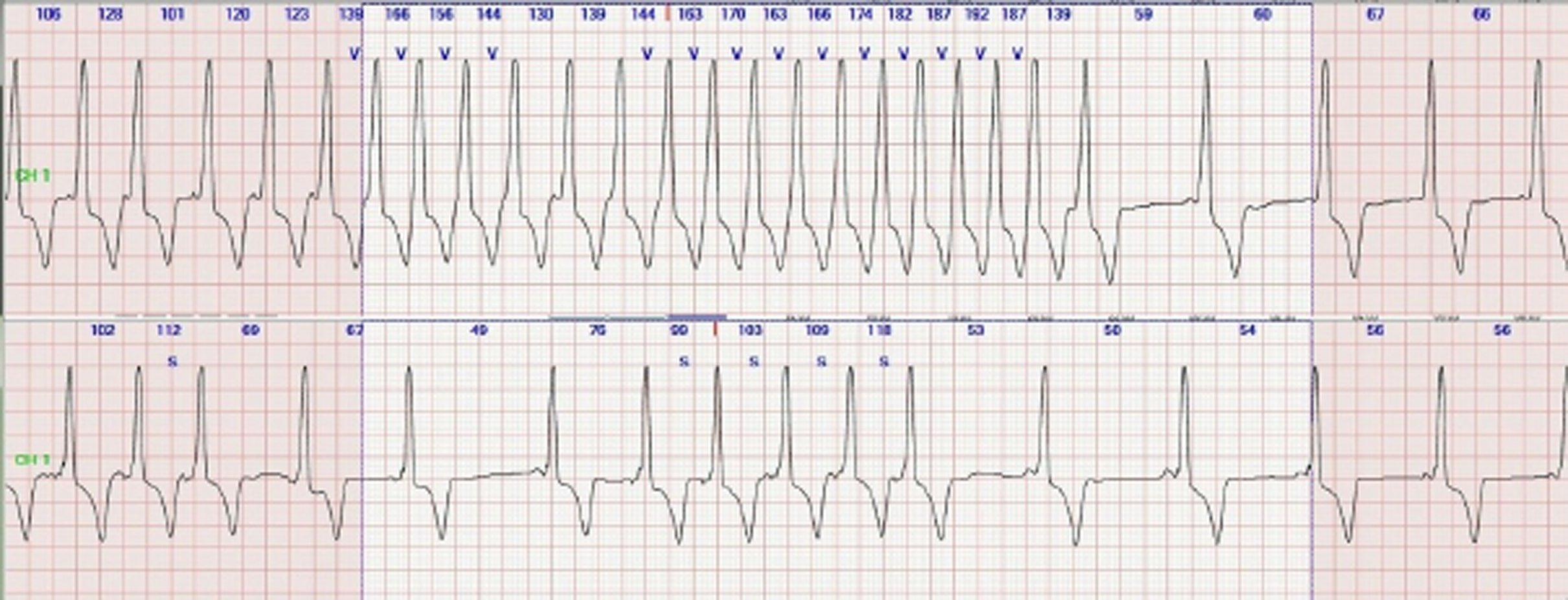
Figure 2. The patient’s basal rhythm was compatible with Wolff-Parkinson-White pattern, and recurrent non-sustained VT (top) SVT (bottom) attacks are detected during the patient’s 24-hour Holter monitoring.
Discussion
Danon disease was first identified in two males with myopathy, cardiomyopathy, and mental retardation in 1981 as “lysosomal glycogen storage disease with normal acid maltase activity,” also called Glycogen Storage Disease Type 2b. Reference Danon, Oh and DiMauro1 It was later discovered that it is caused by the LAMP-2 mutation, which results in a lack of lysosome-associated membrane protein that is associated in intracellular waste and senescent organelle autophagy. Reference Cenacchi, Papa, Pegoraro, Marozzo, Fanin and Angelini2 Accumulation of intracytoplasmic vacuoles containing autophagic material and glycogen in skeletal muscle and myocardial cells constitutes the basic pathophysiology of the disease. The LAMP-2 mutation has been found in 4%–6% of children with hypertrophic cardiomyopathies. Reference Cenacchi, Papa, Pegoraro, Marozzo, Fanin and Angelini2,Reference Hayashi, Tanimoto and Hirayama-Yamada3
Danon disease is a multisystemic disease with a classic triad of cardiomyopathy, myopathy, and mental retardation. There may also be involvement of the eyes, liver, and, in rare cases, the lungs. Symptoms in males occur in early childhood and in adolescence and young adulthood in females. Reference Boucek, Jirikowic and Taylor4 While the earliest symptom onset age reported in the literature was 4 months, the age at diagnosis range from 0 to 65 years. Reference Bertini, Donati and Broda5,Reference Lotan, Salazar-Mendiguchía and Mogensen6 While our first case was asymptomatic, second case was found to be symptomatic for the first time when she was 16 years old.
Almost all patients have cardiac involvement and present with fatigue, chest pain, palpitations, or syncope. While hypertrophic cardiomyopathy is seen in 96% of males, dilated cardiomyopathy can be seen in 27%–29% of females. Reference Brambatti, Caspi and Maolo7 Biventricular hypertrophic cardiomyopathy was evident in our cases. In addition, as a result of diffuse hypertrophy, fibrosis and scar tissue formation, many types of arrhythmias can be seen, such as conduction disorders, accessory pathways, and atrial and ventricular arrhythmias. Wolff-Parkinson-White (WPW) pattern and pre-excitation due to fasciculoventricular pathways are seen in 33%–69% of male patients and 27%–43% of females. Reference Lotan, Salazar-Mendiguchía and Mogensen6,Reference Jhaveri, Herber, Zahka, Boyle, Saarel and Aziz8 In our cases, Wolff-Parkinson-White pattern was also evident on electrocardiogram, and in our second case, ventricular tachycardia attacks were also present. Skeletal myopathy manifests as proximal muscle weakness, myalgia, and limited exercise tolerance. The creatinine kinase elevation seen in most patients was also present in our cases.
Danon disease should be considered in every patient with hypertrophic cardiomyopathy, skeletal myopathy, and neurocognitive findings. Electrocardiography, liver transaminase, serum creatinine kinase, lactate dehydrogenase, brain natriuretic peptide, and troponin levels should be examined in cases of clinical suspicion. Arrhythmias should be screened with 24-hour rhythm Holter monitoring in the presence of normal electrocardiography results. Electromyography supports the diagnosis of myopathy. Echocardiography is the gold standard for the diagnosis of hypertrophic or dilated cardiomyopathy. The use of cardiac MRI is increasing in the evaluation of myocardial scarring, oedema, and fibrosis. Reference Rigolli, Kahn, Brambatti, Contijoch and Adler9 Definitive diagnosis can be made by mutation analysis for LAMP-2 gene.
The major goals of treatment are to alleviate heart failure, prevent sudden death, and avoid neurological impairment. Diuretic and calcium channel blockers that have the potential to cause hypovolemia should be avoided, and competitive sports and heavy exercise should be forbidden. The American Heart Association recommends implantable cardioverter defibrillator implantation for patients with a history of cardiac arrest, sustained ventricular tachycardia, or ventricular fibrillation. Reference Ommen, Mital and Burke10 Implantable cardioverter defibrillator implantation was recommended to our female patient, but her family did not approve. Despite all these approaches, heart failure is progressive and death is inevitable if cardiac transplantation is not performed. Genetic counselling is recommended for all families in Danon disease. Studies on autophagy-modifying agents and gene therapy are still ongoing.
Conclusion
Danon disease is a multisystemic disease that should be considered in every patient with hypertrophic cardiomyopathy, skeletal myopathy, and neurocognitive findings. Progressive cardiac failure and arrhythmias are the causes of death. Mortality can be minimised by implantable cardioverter defibrillator implantation and cardiac transplantation at the appropriate time.
Acknowledgements
None.
Financial support
This research received no specific grant from any funding agency, commercial, or not-for-profit sectors.
Conflicts of interest
None.
Ethical standards
The authors declare that all the procedures that contributed to this work were carried out in compliance with the ethical standards of the relevant national guidelines on human experimentation and with the Helsinki Declaration of 1975, as revised in 2008.



 Open access
Open access