


- HC
haptocorrin
- IF
intrinsic factor
- MCV
mean corpuscular volume
- MMA
methylmalonic acid
- NTD
neural-tube defects
- PA
pernicious anaemia
- PGA
pteroylglutamic acid
- TC
transcobalamin
- THF
tetrahydrofolate
Interaction between folate and vitamin B12
There is no doubt that the debate surrounding the fortification of flour with folic acid is not without controversy. One of the most controversial issues is the assertion that folic acid may mask vitamin B12 deficiency in the elderly. In order to appreciate some key aspects of this controversy an understanding of the metabolic inter-relationship between folic acid and vitamin B12 is required. (Pteroylglutamic acid (PGA) is the form of folate used in both supplements and fortified foods containing folic acid. It is not normally found naturally occurring in foods. The use of the term ‘folic acid’ throughout the present article refers specifically to PGA.) Metabolic functions of folate and vitamin B12 intersect in the methylation cycle. One of the vitamin B12-dependent enzymes (methionine synthase) functions in one of the folate cycles, i.e. the methylation cycle (see Fig. 1). This cycle is necessary to maintain the supply of the methyl donor S-adenosylmethionine, and if it is interrupted it results in a reduction in a wide range of methylated products. One such important methylation process is that of myelin basic protein; its reduction, as is seen in pernicious anaemia (PA) and other causes of vitamin B12 deficiency, produces sub-acute combined degeneration of the spinal cord. Methylation defects are postulated to be the likely cause of myelin damage and disturbed neurotransmitter metabolism(Reference Reynolds1). Sub-acute combined degeneration of the spinal cord is one of the main presenting conditions in PA. The other principal presenting condition in PA is a megaloblastic anaemia morphologically identical to that seen in folate deficiency. The relationship between impairment of the methylation cycle and a lack of DNA biosynthesis and anaemia is explained by ‘the methyl trap hypothesis’(Reference Scott and Weir2). This hypothesis suggests that when the folate cofactor 5-methyltetrahydofolate (THF) is formed, the enzyme 5,10-methyleneTHF reductase that forms the cofactor cannot use it in the back reaction in vivo. Thus, the only way for this folate cofactor to be recycled to THF (and thus participate in DNA biosynthesis and allow cell division), is through the vitamin B12-dependent enzyme methionine synthase (see Fig. 1). When the activity of the enzyme is compromized, as it would be in PA, the cellular folates will become progressively trapped as 5-methylTHF. As a result, the cell will suffer a form of folate deficiency, in that it will have adequate folate but it will be trapped in a form that cannot be used for DNA biosynthesis. The consequence will be an anaemia identical to that seen in true folate deficiency. Thus, in PA there will be a neuropathy and/or anaemia(Reference Scott, Dinn, Wilson and Weir3). Treatment with vitamin B12, if given intramuscularly, will reactivate methionine synthase, allowing myelination to restart. The trapped folate will also be released, thus allowing DNA synthesis and generation of erythrocytes, redressing the anaemia. It is known that treatment with high concentrations of folic acid will also treat the anaemia but not the neuropathy in PA, because when the synthetic form of folate (folic acid) enters the cell it is first converted via dihydrofolate to THF (step ‘X’ in Fig. 1). This process initially occurs outside the methyl trap and can thus initiate DNA biosynthesis, so treating the anaemia. While such folic acid is ultimately trapped as 5-methylTHF, if it is given continuously and in amounts high enough to exceed the capacity of the intestinal mucosal cells to convert it to 5-methylTHF, the folic acid will keep DNA biosynthesis going and thus appear to have treated the anaemia. However, administration of folic acid cannot do anything to restart the methylation cycle, blocked as it is at the vitamin B12-dependent enzyme methionine synthase. Thus, the neuropathy will continue, only to be diagnosed later when treatment with vitamin B12 may only partly reverse the nerve damage. This aspect is discussed later (p. 554).

Fig. 1. Methylation cycle. DOPA, l-3,4-dihydroxyphenylalanine. (Adapted from Scott(Reference Scott, Garrow, James and Ralph85).)
In addition to causing neuropathy and anaemia, even a modest reduction in vitamin B12 status will cause elevation of plasma homocysteine because one of the principal routes of homocysteine metabolism is to recycle homocysteine to methionine via methionine synthase (see Fig. 1).
Normal and abnormal vitamin B12 absorption
The impact of folate and vitamin B12 deficiencies in the elderly cannot be underestimated. Among older participants of the National Health and Nutrition Examination Survey III 14% of anaemia was caused by folate and/or vitamin B12 deficiencies(Reference Woodman, Ferrucci and Guralnik4). A further 20% were caused by Fe deficiency combined with either folate or vitamin B12 deficiency. Thus, folate and/or vitamin B12 deficiencies are the cause of approximately one-third (34%) of cases of anaemia in the USA. Vitamin B12 deficiency is more common among the elderly than among younger adults. This higher prevalence can be more fully understood by examining the mechanism by which dietary vitamin B12 is absorbed. As Fig. 2 illustrates, vitamin B12 is released from food within the lumen of the stomach when it is exposed to gastric acid. Once liberated, vitamin B12 binds with high affinity to haptocorrin (HC; also known as R protein), which is primarily produced by the salivary glands. Since HC has a high binding affinity at acidic pH, most dietary vitamin B12 remains bound to HC in the stomach. Gastric juice containing the vitamin B12–HC complex is then emptied into the duodenum along with intrinsic factor (IF), which is secreted by stimulated parietal cells. In the small intestine vitamin B12 is released from the HC when it comes into contact with bicarbonate, which along with the pancreatic proteases hydrolyses the HC. The free vitamin B12 then binds to IF and the vitamin B12–IF complex moves within the intestinal fluid to the terminal ileum. In the ileum the vitamin B12–IF complex binds to specific receptors present on some enterocytes (a process that can take place in the presence of Ca and a neutral pH). Endocytosis follows and, once inside the cell, IF is degraded and the vitamin B12 transferred to the mitochondria. Free vitamin B12 is then transferred to secretory vesicles in the basal portion of the ileal enterocyte, where it binds to transcobalamin (TC) II. The vitamin B12–TC II complex is liberated from the ileal enterocyte and enters the portal circulation for storage in the liver or release into the plasma for use by tissues(Reference Schjonsby5, Reference Chang, Sitrin and Black6).
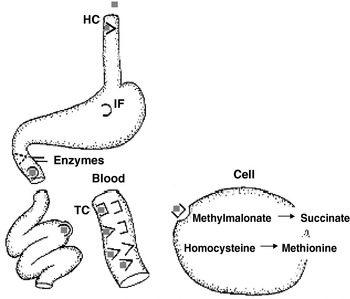
Fig. 2. Absorption of vitamin B12 in human subjects. ( ![]() ), Cobalamin in food; (
), Cobalamin in food; (![]() ), haptocorrin (HC; also termed R protein); (⋂), intrinsic factor (IF); (⊐), transcobalamin (TC). For a detailed explanation of the absorption process, see p. 550.
), haptocorrin (HC; also termed R protein); (⋂), intrinsic factor (IF); (⊐), transcobalamin (TC). For a detailed explanation of the absorption process, see p. 550.
Studies have suggested that an alternative route of vitamin B12 absorption occurs when large doses are taken orally (1000–2000 μg)(Reference Baik and Russell7). This relatively inefficient route of absorption probably occurs by means of passive diffusion throughout the length of the small intestine(Reference Berlin, Berlin and Brante8). It is estimated that only approximately 1% of the administered dose is absorbed by this route(Reference Baik and Russell7). However, it is argued that, in addition to folic acid, fortification of flour with large doses of vitamin B12 might facilitate raising vitamin B12 status in the elderly by passive diffusion and thus allaying fears of undetected vitamin B12 deficiency. This issue is discussed later (p. 556).
Vitamin B12 deficiency in the elderly
Causes
In the elderly adequate provision of vitamin B12 may be limited by dietary supply or more commonly when one or more steps in normal absorption, as described earlier, are affected by disease(s). Malabsorption of cobalamin becomes more frequent with age. In elderly patients vitamin B12 deficiency is caused primarily by inadequate production of IF causing malabsorption of food cobalamin and PA. It also occurs in pancreatic insufficiency, because of a lack of the enzymes needed to liberate cobalamin from the cobalamin–HC complex(Reference Nexo, Hansen, Rasmussen, Lindgren and Grasbeck9). Cobalamin deficiency caused by dietary deficiency or malabsorption (after release of cobalamin from food) is rarer(Reference Matthews10, Reference Pautas, Cherin, de Jaeger and Godeau11). In American studies in which a total of >200 patients with a proven cobalamin deficiency were followed malabsorption of food cobalamin was found to account for about 60–70% of the cases among elderly patients and PA to account for 15–20% of the cases(Reference Andres, Goichot and Schlienger12–Reference Loukili, Noel, Blaison, Goichot, Kaltenbach, Rondeau and Andres15). Other causes included dietary deficiency (<5%), malabsorption (<5%), and hereditary cobalamin-metabolism diseases (<1%).
First described in 1995(Reference Carmel16), food-cobalamin malabsorption syndrome is characterized by the inability to release cobalamin from food or from intestinal transport proteins, particularly in the presence of hypochlorhydria in which the absorption of ‘unbound’ cobalamin remains normal. Malabsorption of food cobalamin is caused primarily by gastric atrophy. Of patients >80 years >40% have gastric atrophy that may or may not be related to Helicobacter pylori infection(Reference Pautas, Cherin, de Jaeger and Godeau11, Reference Carmel, Aurangzeb and Qian17). Other factors that contribute to malabsorption of food cobalamin in the elderly include chronic carriage of H. pylori and intestinal microbial proliferation, which can be caused by antibiotic treatment(Reference Kaptan, Beyan, Ural, Cetin, Avcu, Gulsen, Finci and Yalcin18), long-term ingestion of biguandies (metformin)(Reference Bauman, Shaw, Jayatilleke, Spungen and Herbert19, Reference Andres, Noel and Goichot20) and antacids, including H2-receptor antagonists and proton pump inhibitors(Reference Howden21, Reference Andres, Noel and Abdelghani22); the latter being particularly prevalent among patients with Zollinger-Ellison syndrome(Reference Termanini, Gibril, Sutliff, Yu, Venzon and Jensen23), chronic alcoholism, surgery or gastric reconstruction (e.g. bypass surgery for obesity) and partial pancreatic exocrine failure(Reference Andres, Perrin, Demangeat, Kurtz, Vinzio, Grunenberger, Goichot and Schlienger14, Reference Carmel24).
PA is a classic cause of cobalamin deficiency and one of the most frequent among elderly patients; 20–50% of cases according to two studies(Reference Lee, Herbert, Lee, Foerster, Lukens, Paraskevas, Greer and Rodgers25, Reference Pruthi and Tefferi26) and >15% in another patient series(Reference Andres, Goichot and Schlienger12–Reference Loukili, Noel, Blaison, Goichot, Kaltenbach, Rondeau and Andres15). PA is an autoimmune disease characterized by the destruction of the gastric mucosa, by a primarily cell-mediated process(Reference Toh, van Driel and Gleeson27). Gastric secretions are neutral to slightly acidic even in the presence of gastrin (which normally increases acidity) and contain little or no IF(Reference Lee, Herbert, Lee, Foerster, Lukens, Paraskevas, Greer and Rodgers25, Reference Toh, van Driel and Gleeson27, Reference Nicolas and Gueant28).
Inadequate intake or nutritional deficiency of cobalamin is rare among healthy adults in industrialized countries, even among elderly adults; <5% according to one report(Reference Andres, Perrin, Demangeat, Kurtz, Vinzio, Grunenberger, Goichot and Schlienger14). It is limited to rare instances of patients on strict vegetarian diets and those who are already malnourished, such as elderly patients, patients in institutions or patients in psychiatric hospitals(Reference Herbert29, Reference Russell30). US studies of the dietary intake of the elderly have shown that ⩽50% may have an insufficient intake of cobalamin (<2 μg/d)(Reference Russell30). Such studies, however, are extremely difficult to conduct because they rely mainly on dietary histories(Reference Andres, Perrin, Kraemer, Goichot, Demengeat, Ruellan, Grunenberger, Constantinesco and Schlienger31). Moreover, even if present, dietary deficiency does not result in symptomatic cobalamin deficiency until hepatic reserves are exhausted.
Prevalence
Estimates among the elderly vary widely; from 5% (based upon a low serum vitamin B12 concentration) among institutionalized elderly adults in Germany, The Netherlands and Belgium(Reference Joosten, van den Berg, Riezler, Naurath, Lindenbaum, Stabler and Allen32) to 46% (based on multiple criteria including serum vitamin B12 and methylmalonic acid (MMA)) among institutionalized elderly adults in the UK(Reference Bates, Schneede, Mishra, Prentice and Mansoor33). The impact on estimates of prevalence of using different criteria of deficiency can be illustrated by adapting data presented for a UK cohort of elderly adults(Reference Clarke, Refsum and Birks34). Deficiency may be defined as having a serum vitamin B12 concentration below a cut-off (defined as frankly deficient) or it may be defined further to include those with borderline serum vitamin B12 and accompanying elevated levels of MMA and/or homocysteine or as simply having borderline serum vitamin B12 alone. When this analysis was carried out on the data from the UK cohort(Reference Clarke, Refsum and Birks34) inadequate status, including those with borderline serum vitamin B12, was indicated among males; approximately one-quarter of 65–74 year olds and more than one-third of >75 year olds could be defined as having a compromized status (see Fig. 3).

Fig. 3. Variation in the interpretation of the incidence (%) of vitamin B12 deficiency dependent on the criteria used (adapted from data for a UK cohort of elderly adults(Reference Clarke, Refsum and Birks34)). Criterion A (///), serum vitamin B12 <150 pm; criterion B (![]() ), serum vitamin B12 150–200 pm and elevated methylmalonic acid (MMA; >0·35 μm) and elevated homocysteine (>15 μm); criterion C (■), serum vitamin B12 150–200 pm and elevated MMA (>0·35 μm) but normal homocysteine (<15 μm); criterion D (□), serum vitamin B12 150–200 pm and elevated homocysteine (>15 μm) but normal MMA (<0·35 μm); criterion E (
), serum vitamin B12 150–200 pm and elevated methylmalonic acid (MMA; >0·35 μm) and elevated homocysteine (>15 μm); criterion C (■), serum vitamin B12 150–200 pm and elevated MMA (>0·35 μm) but normal homocysteine (<15 μm); criterion D (□), serum vitamin B12 150–200 pm and elevated homocysteine (>15 μm) but normal MMA (<0·35 μm); criterion E (![]() ), serum vitamin B12 150–200 pm and normal MMA and homocysteine.
), serum vitamin B12 150–200 pm and normal MMA and homocysteine.
Clinical symptoms and diagnosis
Cobalamin deficiency can take years to develop(Reference Savage and Lindenbaum35). Consequently, the clinical symptoms of ‘classical’ cobalamin deficiency, i.e. of severe megaloblastic anaemia combined with neuropsychiatric symptoms, are rarely seen today(Reference Stabler and Allen36). However, vitamin B12 deficiency often goes unrecognized because the initial clinical manifestations are subtle and non-specific or are attributed to the normal ageing process(Reference Carmel37).
Detection and treatment of the early stages of cobalamin deficiency is important(Reference Bjorkegren and Svardsudd38), particularly in patients with neuropsychiatric symptoms because they may fail to respond if therapy is delayed(Reference Healton, Savage, Brust, Garrett and Lindenbaum39). All patients with unexplained anaemia and/or unexplained neuropsychiatric symptoms should ideally be examined for cobalamin deficiency, because vitamin B12 deficiency can result in neurological symptoms without haematological abnormalities. An inverse relationship between the presence of neurological and haematological disease in vitamin B12 deficiency has been reported(Reference Stabler40); the higher the packed cell volume, the worse the neuropathy. The early stages of vitamin B12 deficiency will not lead to macrocytosis or anaemia, because biochemical vitamin B12 deficit precedes overt clinical deficiency with macro-ovalocytes and falling Hb levels. Macrocytosis can be blocked by concurrent Fe deficiency. The sensitivity of mean corpuscular volume (MCV) to detect vitamin B12 deficiency is low, and many patients with vitamin B12 deficiency will remain unnoticed when the MCV is used to decide whether further evaluation is necessary (see p. 552).
Haematological signs, especially macrocytic anaemia, have traditionally been the cornerstone of diagnostics for cobalamin status(Reference Chanarin and Metz41), but this position has been re-evaluated during the last few years(Reference Snow42). In a review of the sequelae of symptoms of vitamin B12 deficiency, it was commented that anaemia is not present in all cases of vitamin B12 deficiency and discrete neuropsychiatric symptoms are often the only signs(Reference Nexo, Hansen, Rasmussen, Lindgren and Grasbeck9). The concept that patients suffering from cobalamin deficiency may have neuropsychiatric symptoms without macrocytosis or anaemia was not widely accepted until relatively recently(Reference Lindenbaum, Healton, Savage, Brust, Garrett, Podell, Marcell, Stabler and Allen43). Even though macrocytosis is considered a diagnostic, increased MCV values are in most cases related to other conditions such as drugs and alcohol abuse. Cobalamin and folate deficiencies have been shown to be responsible for macrocytosis in only about 6% of cases(Reference Savage, Ogundipe, Allen, Stabler and Lindenbaum44). Macrocytic anaemia is therefore not mandatory. Normocytic anaemia is seen if the patient suffers from both cobalamin and Fe deficiencies. Anaemia is seen in approximately 60% of patients with cobalamin deficiency and neuropsychiatric symptoms without anaemia are seen in ⩽40% of patients(Reference Nexo, Hansen, Rasmussen, Lindgren and Grasbeck9). Thus, it has been realized that neuropsychiatric illness may be the only symptom of cobalamin deficiency and, importantly, the neurological symptoms are considered of most concern because they may fail to remit on therapy. Of patients with neuropsychiatric illness approximately 80% have neurological symptoms, while 10% have psychiatric symptoms.
Stages of vitamin B12 deficiency
It is now commonly accepted that there are five stages of vitamin B12 deficiency that between them encompass the range from normal status to severe deficiency(Reference Herbert45). The earliest marker of a negative vitamin B12 balance is probably a low holoTC II concentration (<40 pg/ml). As the deficiency progresses at least one of the metabolic markers (most commonly MMA or less often homocysteine) will also begin to rise as vitamin B12 becomes unavailable for the associated chemical reactions. Further progression will lead to an elevated MCV and, eventually, outright anaemia.
Importantly, it is still not clear at which stage neurological damage first appears during decreasing vitamin B12 status. It is therefore not yet known when vitamin B12 deficiency becomes clinically important or, alternatively, when the damage caused by such deficiency first occurs (as soon as levels start to fall or as deficiency progresses). For example, is neurological impairment reversible in patients with decreased holoTC levels but normal levels of both MMA and homocysteine? What is clear is that both MCV and Hb reach levels that would be deemed clinically abnormal only when vitamin B12 deficiency reaches a very advanced stage. In clinical practice vitamin B12 testing is often limited to patients who have an elevated MCV, macrocytic anaemia or specific neurological symptoms(Reference Carmel46).
Mean corpuscular volume as a diagnostic marker
The use of any marker as a diagnostic test for the detection or exclusion of vitamin B12 deficiency is only useful if its diagnostic accuracy is sufficiently high. For MCV to be a good marker of vitamin B12 deficiency it would be expected that the low serum vitamin B12 level would have a corresponding elevated MCV. However, according to data from the National Diet and Nutrition Survey of those aged >65 years(Reference Finch, Doyle, Lowe, Bates, Prentice, Smithers and Clarke47) there is a marked discrepancy between the numbers of the elderly who have low serum vitamin B12 levels and numbers of those who have elevated MCV(48).
MCV should not be relied on when screening for vitamin B12 deficiency(Reference Thompson, Babitz, Cassino, Freedman and Lipkin49, Reference Thompson, Cassino, Babitz, Meola, Berman, Lipkin and Freedman50). A systematic review has shown that the MCV is far from perfect in terms of detecting or excluding vitamin B12 deficiency, both for patients with anaemia and those without anaemia(Reference Oosterhuis, Niessen, Bossuyt, Sanders and Sturk51). In fact, a considerable number of patients who are vitamin B12 deficient will remain unnoticed when the MCV is used to decide which patients require further evaluation. Depending on the population studied, ⩽84% of cases may be missed. Thus, the positive predictive value of elevated MCV is low, whereas the negative predictive value is considered rather high(Reference Oosterhuis, Niessen, Bossuyt, Sanders and Sturk51). Another study has concluded that concomitant evaluation of erythrocyte distribution width could improve the diagnostic specificity of MCV(Reference Bessman, Gilmer and Gardner52). Of the patients with an elevated MCV ⩽55% will show a vitamin B12 deficiency on further testing(Reference Oosterhuis, Niessen, Bossuyt, Sanders and Sturk51). Thus, MCV can be used to diagnose whether vitamin B12 deficiency is more probable or less probable but cannot be any more accurate. An elevated MCV justifies the measurement of serum vitamin B12 and should not be the only criterion used to rule out a diagnosis of vitamin B12 deficiency.
Haematological Markers and Vitamin B12 Deficiency in the Elderly Study
The Haematological Markers and Vitamin B12 Deficiency in the Elderly Study has confirmed previous findings that MCV is not a good predictor of vitamin B12 status(Reference Mooney, Young, Patterson and Cuskelly53). The objective of the study was to assess the prevalence of abnormal haematological markers among hospitalized subjects with low serum vitamin B12 levels. In this analysis serum folate and serum vitamin B12 were collated from all subjects aged 65–85 years, who had these measurements taken in a 6-month period (February–July 2003). Serum ferritin, Hb, MCV and packed cell volume were included if they had been measured within ±4 d of serum folate and vitamin B12 measurements. Data were divided into quintiles of either serum folate or serum vitamin B12 and the prevalence of anaemia was compared between quintiles. The analysis shows that when subjects are stratified (into quintiles) according to vitamin B12 status, MCV is as likely to be elevated among those subjects with normal serum vitamin B12 levels as it is among those with low serum vitamin B12 levels (Fig. 4).

Fig. 4. Incidence of high mean corpuscular volume (MCV) and low Hb in subjects aged 65–85 years participating in the Haematological Markers and Vitamin B12 Deficiency in the Elderly Study(Reference Mooney, Young, Patterson and Cuskelly53). (A) Incidence of high MCV and low Hb by quintile of serum vitamin B12. (□), Percentage with high MCV; (///), serum vitamin B12 (ng/l×10−1); (■), percentage with low Hb. (B) Incidence of high MCV and low Hb by quintile of serum folate(Reference Mooney, Young, Patterson and Cuskelly53). (□), Percentage with high MCV; (///), serum folate (μg/l×10) (■), percentage with low Hb.
Biological markers of vitamin B12 status
It is now possible to diagnose early stages of deficiency, even before the development of clinical symptoms(Reference Schneede and Ueland54), as a result of the development of more specific and sensitive diagnostic tests for cobalamin deficiency that can also identify atypical forms of deficiency(Reference Carmel55). However, accompanying the advent of these diagnostic tests is the risk of their inappropriate use without reference to clinical signs and symptoms. The advisability of using this approach has been questioned and there certainly is a balance to be struck between the risks of under-recognition of vitamin B12 deficiency and potential over-diagnosis and over-treatment of the disease(Reference Chanarin and Metz41).
The definition of, or diagnosis of, a disease based purely on abnormal biochemical variables is, however, problematic(Reference Chanarin and Metz41) and invokes the danger of circular reasoning, especially if the diagnostic approach no longer includes clinical criteria(Reference Schneede and Ueland54).
Serum cobalamin
The diagnostic utility of serum total cobalamin has been questioned because total cobalamin in blood may not accurately reflect intracellular vitamin B12 status in the different tissues. As a result of the lack of specificity, the lower cut-off limits for serum cobalamin have been reduced at the expense of sensitivity(Reference Carmel, Green, Rosenblatt and Watkins56). As a result, total cobalamin in serum is now considered an unreliable indicator of functional cobalamin status(Reference Joosten, van den Berg, Riezler, Naurath, Lindenbaum, Stabler and Allen32, Reference Lindenbaum, Savage, Stabler and Allen57).
Furthermore, cobalamin may be strongly influenced by folate deficiency(Reference Ward58). There are several conditions under which artificially-high or -low total vitamin B12 concentrations can be measured, which greatly affect the specificity of the serum vitamin B12 test in these patients(Reference Schneede and Ueland54). These conditions include: falsely-elevated serum vitamin B12 caused by liver disease, myeloproliferative disease, TC II deficiency, small intestinal bacterial overgrowth and haemolysis; falsely-low serum vitamin B12 caused by severe folate deficiency, severe Fe deficiency, oral contraceptives, mild or severe HC deficiency and myeloma.
Holotranscobalamin
Most cobalamin in serum is bound to HC. Only a minor percentage, 20–30, is bound to TC, which is essential for the transport of cobalamin from the ileum to the majority of tissues and for cellular uptake through a receptor-mediated process involving a specific receptor TC-R(Reference Bose, Seetharam and Seetharam59). HoloTC may be a better marker of recently-absorbed cobalamin(Reference Bor, Nexo and Hvas60) and has been considered a more sensitive marker of cobalamin status than total cobalamin(Reference Hvas and Nexo61).
Factors other than cobalamin status, however, may affect the holoTC levels, and possibly its relationship with the metabolic markers. These factors include a common genetic polymorphism 776 (C776G) of the TC gene and impaired renal function and liver disease(Reference Carmel62–Reference Fodinger, Veitl and Skoupy64), as well as female sex hormones(Reference Riedel, Bjorke Monsen, Ueland and Schneede65). The influence on holoTC of factors that are unrelated to cobalamin status may weaken the utility of holoTC as a diagnostic marker. Moreover, the fact that holoTC (like serum cobalamin) cannot be used to monitor response to cobalamin supplementation is another limitation of this marker. Further research is necessary to evaluate the usefulness and limitations of holoTC in clinical practice.
Methylmalonic acid
A lack of vitamin B12 (or more specifically, a lack of hydroxocobalamin) leads to an accumulation of both l-methylmalonyl-CoA and its precursor d-methylmalonyl-CoA. Excess d-methylmalonyl-CoA is then converted into MMA (measurable in both serum and urine). Overall, the sensitivity of increased serum MMA in the diagnosis of vitamin B12 deficiency appears to be greater than that of serum homocysteine(Reference Lindenbaum, Savage, Stabler and Allen57).
However, there are some major problems with the measurement of MMA in terms of its application in clinical practice. Measurement methods (GC–MS and HPLC) are cumbersome and require expensive equipment. MMA may also be increased in renal insufficiency and hypovolaemic states(Reference Savage, Lindenbaum, Stabler and Allen66, Reference Stabler, Marcell, Podell, Allen and Lindenbaum67).
The greatest difficulty with MMA is how it may be interpreted. The prevalence of high serum MMA in elderly patients is substantially higher than either low serum vitamin B12 levels or a clinically-evident deficiency state(Reference Snow42, Reference Pennypacker, Allen, Kelly, Matthews, Grigsby, Kaye, Lindenbaum and Stabler68), which could mean that elevated MMA precedes low serum vitamin B12 levels or that it is more sensitive. It is recommended that elevated MMA be interpreted with caution in the absence of further evidence of a deficiency state(Reference Chanarin and Metz41, Reference Green69).
Masking of vitamin B12 deficiency
The most important benefit of folic acid fortification of staple foods (such as flour) is its ability to optimize folic acid intakes without any need for the consumer to change his/her food intake. Its accompanying disadvantage is the provision of additional folic acid to those who do not need it or who may in fact be in danger as a result of consuming it. Therein lies the argument against folic acid fortification, despite its establishment in USA since 1998 (a strategy to reduce neural-tube defects (NTD)). The USA has chosen to use PGA as the form of folic acid to add to flour and cereals, possibly on the grounds of cost (PGA is markedly cheaper than 5-methylTHF), and while early data suggest that NTD incidence has decreased appreciably (a 19% decrease since fortification was introduced(Reference Honein, Paulozzi, Mathews, Erickson and Wong70)), a number of arguments against fortification have been proposed based on the metabolism of PGA by human subjects. Two specific arguments exist that folic acid: (1) accelerates the neurological damage caused by vitamin B12 deficiency; (2) cures or ‘masks’ the anaemia of vitamin B12 deficiency, thereby hiding or ‘masking’ a sign of deficiency on which clinicians may be dependent for diagnosis.
Vitamin B12 deficiency and folic acid supplementation and accelerated neurological damage
Accepting that vitamin B12 deficiency may only manifest clinically in some patients as neurological damage, it has been suggested that administration of high doses of folic acid might have precipitated neurological degeneration. It is now not possible to prove whether or not there is an increased rate of sub-acute combined degeneration of the spinal cord, or whether the neuropathy is made more severe as a result of giving folic acid to vitamin B12-deficient individuals. Such a study would be unethical. Although folic acid is not toxic to normal individuals even in large doses, it might appear to be neurotoxic in the special circumstances of vitamin B12 deficiency. In general, it is agreed that there are no data that demonstrate that folic acid is harmful to the nervous system in individuals with vitamin B12 deficiency at doses of ⩽1 mg folic acid/d in the short term. However, folic acid can correct the anaemia of vitamin B12 deficiency in some patients even at doses <1 mg/d(48). This outcome could alter the presentation of vitamin B12 deficiency in the population (with a larger proportion of patients presenting with altered sensation and a smaller proportion presenting with symptoms of anaemia). It is not possible to predict the effect on the incidence of irreversible vitamin B12 neuropathy, although arguments can be put forward to suggest it might increase as a result of a delay in the diagnosis, which can depend on detecting anaemia. Any general policy to increase dietary folic acid consumption should require vigilance in relation to the incidence, prevalence and presentation of vitamin B12 neuropathy in the community to determine whether these data change.
Effects of folic acid on reticulocytosis in vitamin B12 deficiency
At high doses (yet to be determined, but in healthy adults possibly >300 μg in a bolus dose(Reference Kelly, McPartlin, Goggins, Weir and Scott71)), the metabolic handling of PGA changes during the absorption process. A non-saturable transport mechanism involving passive diffusion of this unaltered vitamin allows the passage of PGA from the gut, into the portal circulation and into cells (as unaltered PGA). The uptake of the unaltered vitamin may lie at the centre of the argument against fortification. It is well recognized that both 5-methylTHF (the predominant circulating form of folate) and vitamin B12 are required to regenerate THF (the substrate for the formation of 5-methyleneTHF) and subsequently for DNA pyrimidine synthesis (see Fig. 1). Thus, during vitamin B12 deficiency this cascade is inhibited and a megaloblastic anaemia may develop (as a result of inadequate DNA synthesis for erythrocyte cell division); an anaemia that is indistinguishable from that associated with folate deficiency. However, unlike 5-methylTHF, PGA, can bypass the vitamin B12-dependent pathway and is converted to THF by another route (step ‘X’ in Fig. 1). Thus, during vitamin B12 deficiency and in the presence of unaltered PGA substrates are still available for DNA pyrimidine synthesis. Thus, normal reticulocytosis is achieved despite vitamin B12 deficiency. Although it was thought that folic acid might exacerbate vitamin B12 deficiency and its symptoms, it is probably not the case(Reference Bower and Wald72). However, if the diagnosis of vitamin B12 deficiency is dependent on the observation of clinical symptoms of anaemia, or morphological changes in erythrocyte cells, which it often is, then folic acid may ‘mask’ the underlying vitamin B12 deficiency by allowing normal pyrimidine synthesis to progress. Unfortunately, the (irreversible) neurological symptoms would also progress untreated. The recent Department of Health report on folate in health has recommended that folic acid fortification is desirable for the prevention of NTD, but is less emphatic about the risks to the elderly (particularly among the elderly with vitamin B12 deficiency)(48). Subsequently, fortification was not recommended in the UK by the Food Standards Agency(73), citing that ‘further evidence on the impact on older people needs to be considered, including the extent to which it would be possible to put in place effective surveillance for vitamin B12 deficiency’. However, in May 2007 the Food Standards Agency Board agreed unanimously that ‘mandatory fortification’ with folic acid should be introduced, alongside controls on voluntary fortification and advice on the use of supplements. (The Scientific Advisory Committee for Nutrition report underpinning this decision(74) is discussed later (p. 555).)
Few systematic data exist in relation to the level of folic acid intake required to mask vitamin B12 deficiency(Reference Koehler, Pareo-Tubbeh, Romero, Baumgartner and Garry75). The majority of available information relates to early case reports of folic acid therapy (mostly high dose, e.g. ≥5 mg/d) for the treatment of PA(73). In general, the data taken from these reports have suggested that supplementation with ⩽1 mg folic acid/d is safe in this respect (i.e. does not alleviate vitamin B12-associated anaemia in the majority of subjects). The effects of doses between 1 and 5 mg folic acid/d are unclear, whilst supplementation with ≥5 mg folic acid/d is reported to reverse the haematological signs of vitamin B12 deficiency in ≥50% of subjects(Reference Bower and Wald72, Reference Chanarin76, Reference Savage, Lindenbaum and Bailey77).
Current data are therefore equivocal as to the impact of PGA (and dose) on haematological responses, and there are no data to assess the dose–response relationship between PGA intake and haematological responses. Data on the effects of low doses of folic acid in vitamin B12 deficiency are very limited, but suggest that approximately half the patients who are vitamin B12 deficient will show a reticulocytosis after the administration of 0·4 mg folic acid/d(Reference Savage, Lindenbaum and Bailey77). It has been suggested that the upper limit of intake should be set at 1 mg folic acid/d(78).
It should be stressed that so-called ‘masking’ of the anaemia of PA is not thought to occur at concentrations of folate found in food, or at intakes of the synthetic form of folic acid at the usual RDA levels of 200 μg/d or 400 μg/d(Reference Kelly, McPartlin, Goggins, Weir and Scott71). However, ‘masking’ definitely occurs at high concentrations of folic acid (>1000 μg/d). The latter becomes a concern when it is proposed to add synthetic folic acid to the diet by way of fortification of a dietary staple such as flour. The concern is that if folic acid is added to have the desirable effect of reducing NTD (and/or lowering plasma homocysteine), those individuals on high flour intakes could inadvertently become exposed to very high levels of folic acid. Included among these individuals are elderly adults whose timely diagnosis of PA (via presentation as the anaemia) might be delayed. This delay in diagnosis could allow the neuropathy to progress to such an extent that when it is eventually diagnosed it is partly or largely irreversible.
Folic Acid Supplementation in Vitamin B12 Deficiency Study
Monitoring and tracking the effects of folic acid fortification on the masking of anaemia is less easy in a population with access to fortified foods such as the USA. Mandatory folic acid fortification does not exist in the UK, and it therefore provides an ideal environment for carrying out a controlled intervention. The Folic Acid Supplementation in Vitamin B12 Deficiency Study was conducted to assess the impact of folic acid supplementation on the incidence of anaemia in subjects who have borderline vitamin B12 deficiency. In populations such as in Northern Ireland where there is limited voluntary fortification elderly adults who consume fortified foods can be easily identified by screening questionnaire. Those individuals who consumed neither folic acid supplements nor fortified foods qualified to participate in the intervention. The study recruited elderly subjects with borderline vitamin B12 deficiency (who would not usually be referred for vitamin B12 therapy in a clinical setting) and randomly assigned them to receive either placebo or 400 μg folic acid/d for 24 weeks. The primary objective was to assess the effect of supplementation on haematological markers and thereby ascertain whether additional folic acid masks anaemia as defined by MCV. Vitamin B12 and folate status were also monitored throughout the intervention. This study has been completed and its results will provide analysis of the effects on haematological markers (including MCV) of a controlled folic acid intervention in elderly adults with borderline vitamin B12 deficiency.
Potential impact of fortification among the elderly
Modelling of the UK National Diet and Nutrition Survey of the elderly(Reference Finch, Doyle, Lowe, Bates, Prentice, Smithers and Clarke47) has been completed by the Committee on Medical Aspects of Health Policy(48) and it was found that intakes of PGA would be >1 mg/d in <1% of those >50 years (at fortification levels of 2·4 mg/kg). However, these estimates are based on modelling of recorded food intakes that may themselves be underestimated. Hence, the data generated from modelling exercises must be treated with caution as they are probably an estimate of the minimal possible effect. In addition, modelling only estimates the additional planned folic acid intake based on theoretical targets and does not therefore take account of actual additional intakes that would almost certainly be higher as a result of overage. However, in 2006 the Scientific Advisory Committee for Nutrition completed a subsequent modelling exercise of the UK National Diet and Nutrition Survey data(74). Their results indicate that fortification would have a less marked impact on increasing folic acid intakes than that predicted by the Committee on Medical Aspects of Health Policy analysis. The Scientific Advisory Committee for Nutrition attribute higher predictions of the Committee on Medical Aspects of Health Policy to not taking processing losses into account and overestimating the percentage of flour in flour products. The Scientific Advisory Committee for Nutrition conclude from their analysis that mandatory fortification at levels as high as 3·0 mg/kg would in fact reduce the numbers of individuals at risk of consuming >1 mg folic acid/d (if the availability of both voluntary fortification of foods with folic acid and non-prescription folic acid supplements were to discontinue).
Impact of fortification in the USA
Fortification in the USA was estimated to provide an average of an additional 0·1 mg folic acid/d, although published data suggest that in fact actual intakes are twice this amount, at an average additional 0·2 mg/d(Reference Quinlivan and Gregory79, Reference Choumenkovitch, Selhub, Wilson, Rader, Rosenberg and Jacques80). There are virtually no data on the Hb response or risk of developing neuropathy at such doses(Reference Savage, Lindenbaum and Bailey77).
Notwithstanding the lack of data on folic acid dose and haematological response, optimism for the safety of fortification for the elderly in the USA has been proposed on the basis of a recent analysis(Reference Mills, Von Kohorn, Conley, Zeller, Cox, Williamson and Dufour81). This analysis tracked the incidence of anaemia among subjects with low vitamin B12 status from the period pre-fortification until 2 years after mandatory fortification was introduced in the USA. They found no obvious trend towards increased incidence of ‘masked’ anaemia among those with low vitamin B12 status. This analysis has both merits and limitations. Fortunately, results are based on clinical observations and therefore reflect the impact of actual intakes of additional folic acid among the US elderly. They are not therefore dependent on modelling data. However, the tracking and monitoring period included was only 5 years post-fortification and possibly not long enough to measure true long-term trends. While this study is considered by some to be a positive indication for implementation of fortification, caution should be exercised about these conclusions on the basis of the limitations of the analysis.
Folic acid fortification: pros and cons for the elderly
Notwithstanding the potential masking effect of folic acid, there may be some potential benefits of fortification for the elderly, not least the ability for fortification to improve folate status. The majority of the elderly who are folate deficient are not vitamin B12 deficient (approximately 90%(Reference Clarke, Refsum and Birks34)). There is no definitive evidence that folic acid causes accelerated neurological damage. Fortification would deliver folic acid in smaller multiple doses throughout the day and there is no evidence to suggest that folic acid consumed in this way by the elderly would lead to free folic acid accumulating in serum. Finally, where a system exists for efficiently monitoring and screening for vitamin B12 deficiency, the issue of masking would not need to be addressed.
On the other hand, there are some potential detrimental effects of fortification. In the absence of an efficient system for screening the elderly population for vitamin B12 deficiency, the potential for PGA to mask vitamin B12 deficiency and the minimum dose at which deficiency occurs has not yet been defined. Furthermore, there has been no conclusion about whether smaller doses spread across the day are safe. In fact, recent evidence suggests that in some population groups they may not be safe(Reference Sweeney, McPartlin, Weir, Daly and Scott82), as studies in pregnant women have suggested that free folic acid levels in serum are sustained to a greater extent when multiple doses are given throughout the day than when a single dose is given once daily. Evidence that additional folic acid could lower the risk of certain cancers and CVD (diseases that have a high incidence among the elderly) is still equivocal(74).
Vitamin B12 fortification
It has been argued that masking of vitamin B12 deficiency is a red herring and could be simply overcome by simultaneous fortification with vitamin B12. The debate about fortification would be less complicated if combined vitamin B12–folic acid fortification was a solution to the potential effect of masking of vitamin B12 deficiency by folic acid. However, adding vitamin B12 would be an ineffective strategy. In most cases of vitamin B12 deficiency the cause is related to malabsorption(74), and therefore oral vitamin B12 would not help to improve status. Even if a large dose was given only 1% of oral vitamin B12 could be absorbed by passive diffusion. High doses of vitamin B12 also have the effect of turning flour yellow. The dose of vitamin B12 that would need to be added would therefore be prohibitive.
Treatment and conclusions
There is a danger that excitement about the benefits of folic acid fortification to one subset of the population (babies at risk of NTD) may obscure the clear necessity to assess its possible drawbacks to another population group (the elderly). Folic acid fortification is undoubtedly a way forward to prevent NTD, but only if its implementation also assumes the responsibility to screen for individuals at risk of vitamin B12 deficiency.
Currently, there is neither an independent gold standard nor a reference method for unequivocal characterization of vitamin B12 deficiency(Reference Carmel24). Moreover, there is no agreement on how to treat (in relation to formulation, dosage, cobalamin preparation, intensity and duration of treatment), or on when and how to monitor the effects of treatment(Reference Schneede83).
It has been suggested that it may be possible to categorize ‘at-risk’ elderly for vitamin B12 deficiency using haematological variables; measured routinely in clinical practice. However, when this hypothesis was tested using clinical data from a UK hospital, it was found that regardless of the comparisons made (>70 years old v. <70 years old; anaemic v. non-anaemic, macrocytosis v. normocytosis; macrocytosis v. microcytosis; normocytosis v. microcytosis) there was no relationship between the prevalence of abnormal haematology and vitamin B12 deficiency(Reference Chui, Lau and Wong84).
In principle, to prevent irreversible neurological damage it may be justifiable to use prophylactic cobalamin treatment only on the basis of early biochemical signs. This approach poses particular demands on the specificity and sensitivity of diagnostic tests(Reference Carmel, Green, Rosenblatt and Watkins56). Some neurological symptoms may become irreversible 1 year after the first clinical signs(Reference Schneede and Ueland54).
Conclusions and recommendations
As long as there still remains a lack of a reliable clinical test for the diagnosis of true vitamin B12 deficiency, any fortification programme should be accompanied by appropriate systems to ensure that the elderly at risk of vitamin B12 deficiency are identified. As concluded by Scientific Advisory Committee for Nutrition, the issue of overage needs to be given careful consideration(74). This approach would appear to be the most balanced way forward.




