



1. Introduction
Adhesion between soft fluctuating surfaces is found in a range of engineering applications and in biological systems. Complex lifeforms rely on cells adhering to each other, facilitated by the binding of membrane-anchored adhesive molecules across the gap between the membranes of the cells. The dynamics of adhesion involves a rich interplay of membrane deformation, chemical kinetics of the aforementioned molecular binding, as well as fluid flow in the narrow space between the membranes, which can give rise to intricate dynamical processes such as the coarsening of adhesion patches. A number of essential physiological processes depend on adhesion dynamics, such as cadherin-mediated adhesion (van Roy & Berx Reference van Roy and Berx2008), lumen formation between cells in a growing embryo (Dumortier et al. Reference Dumortier, Le Verge-Serandour, Tortorelli, Mielke, de Plater, Turlier and Maître2019) and the immune synapse which facilitates an immunological response (Grakoui et al. Reference Grakoui, Bromley, Sumen, Davis, Shaw, Allen and Dustin1999; Dustin & Cooper Reference Dustin and Cooper2000). Experiments and numerical results have shown that the features of these phenomena can be passive processes only relying on the physical forces involved (Qi, Groves & Chakraborty Reference Qi, Groves and Chakraborty2001; Dustin Reference Dustin2010; Le Verge-Serandour & Turlier Reference Le Verge-Serandour and Turlier2021).
One way to describe the adhesion of membranes is to adopt the Helfrich model with an adhesion potential (Seifert & Lipowsky Reference Seifert and Lipowsky1990; Smith et al. Reference Smith, Sackmann and Seifert2003, Reference Smith, Sengupta, Goennenwein, Seifert and Sackmann2008; Agrawal Reference Agrawal2011; Hill & Al-Amodi Reference Hill and Al-Amodi2024). This approach successfully predicts the equilibrium shape of the membrane, but neglects the motion of the extracellular fluid in the cleft between the two membranes. In such nanometrically thin but micrometrically wide channels, however, the forces required to squeeze the viscous extracellular fluid are not negligible, motivating a viscous thin film description of the flow (Leong & Chiam Reference Leong and Chiam2010). The lubrication theory allows for a relatively simple inclusion of forces due to membrane deformation, molecule/protein binding, as well as thermal fluctuations (Carlson & Mahadevan Reference Carlson and Mahadevan2015a
,
Reference Carlson and Mahadevanb
). Membranes resist lateral deformation due to membrane tension
 $\gamma$
(Evans, Waugh & Melnik Reference Evans, Waugh and Melnik1976), as it increases their free energy in a manner analogous to interfacial surface tension, whose influence on the dynamics of thin liquid films has been studied extensively (Oron, Davis & Bankoff Reference Oron, Davis and Bankoff1997; Craster & Matar Reference Craster and Matar2009), particularly in the case of the dewetting of nanoscale thin liquid films (Zhang & Lister Reference Zhang and Lister1999; Mecke & Rauscher Reference Mecke and Rauscher2005; Grün et al. Reference Grün, Mecke and Rauscher2006; Nguyen et al. Reference Nguyen, Fuentes-Cabrera, Fowlkes and Rack2014; Zhang, Sprittles & Lockerby Reference Zhang, Sprittles and Lockerby2019; Zhao et al. Reference Zhao, Liu, Lockerby and Sprittles2022). In addition, membranes of finite width
$\gamma$
(Evans, Waugh & Melnik Reference Evans, Waugh and Melnik1976), as it increases their free energy in a manner analogous to interfacial surface tension, whose influence on the dynamics of thin liquid films has been studied extensively (Oron, Davis & Bankoff Reference Oron, Davis and Bankoff1997; Craster & Matar Reference Craster and Matar2009), particularly in the case of the dewetting of nanoscale thin liquid films (Zhang & Lister Reference Zhang and Lister1999; Mecke & Rauscher Reference Mecke and Rauscher2005; Grün et al. Reference Grün, Mecke and Rauscher2006; Nguyen et al. Reference Nguyen, Fuentes-Cabrera, Fowlkes and Rack2014; Zhang, Sprittles & Lockerby Reference Zhang, Sprittles and Lockerby2019; Zhao et al. Reference Zhao, Liu, Lockerby and Sprittles2022). In addition, membranes of finite width
 $d$
resist bending (Evans Reference Evans1974), as one side is compressed and the other side is stretched, which is characterised by a bending modulus
$d$
resist bending (Evans Reference Evans1974), as one side is compressed and the other side is stretched, which is characterised by a bending modulus
 $B=Ed^3/12(1-\nu ^2)$
, where
$B=Ed^3/12(1-\nu ^2)$
, where
 $E$
is the Young’s modulus and
$E$
is the Young’s modulus and
 $\nu$
is the Poisson ratio. Both tension and bending resist the deformation of a flat membrane, but there are subtle differences in how they act. A bendocapillary length
$\nu$
is the Poisson ratio. Both tension and bending resist the deformation of a flat membrane, but there are subtle differences in how they act. A bendocapillary length
 $l_{BC}=\sqrt {B/\gamma }$
can be obtained by balancing the forces from tension and bending, which is approximately
$l_{BC}=\sqrt {B/\gamma }$
can be obtained by balancing the forces from tension and bending, which is approximately
 $100$
nm for the properties of most cell membranes, and bending dominates for length scales smaller than the critical length scale (Roman & Bico Reference Roman and Bico2010; Deserno Reference Deserno2015).
$100$
nm for the properties of most cell membranes, and bending dominates for length scales smaller than the critical length scale (Roman & Bico Reference Roman and Bico2010; Deserno Reference Deserno2015).
For adhesion to occur, the membranes must first come close enough to each other to allow the adhesive molecules to start to form bonds. In the absence of directed motion due to active cytoskeletal forces or protein–membrane interactions (Liese & Carlson Reference Liese and Carlson2021; Yuan et al. Reference Yuan, Alimohamadi, Bakka, Trementozzi, Day, Fawzi, Rangamani and Stachowiak2021), the forces required to push out the extracellular fluid in the channel can be attributed to thermal fluctuations (Aarts Reference Aarts2004; Delgado-Buscalioni, Chacon & Tarazona Reference Delgado-Buscalioni, Chacon and Tarazona2008) as well as the other fluctuations inherent to living matter (Guo et al. Reference Guo, Ehrlicher, Jensen, Renz, Moore, Goldman, Lippincott-Schwartz, Mackintosh and Weitz2014; Gupta & Guo Reference Gupta and Guo2017). The fluctuations in the width of the channel, although small in amplitude and random in direction, can still bring membranes close enough to initiate adhesive molecule/protein binding if given sufficient time. This process is similar to the spontaneous thermal dewetting of a linearly stable thin viscous film coated on a solid substrate, where thermal fluctuations are needed to bring the liquid free surface close enough to the solid substrate for the disjoining pressure to rupture the film, for which the average waiting time for rupture can be predicted by rare-event theory (Sprittles et al. Reference Sprittles, Liu, Lockerby and Grafke2023; Liu, Sprittles & Grafke Reference Liu, Sprittles and Grafke2024).
After the initial binding of adhesive molecules, the adhesion patches grow in size. If more than one type of adhesive molecule is involved, the adhesion patches can separate into distinct regions (phases) where one specific type of molecule is bound, as is seen in the immune synapse, receptor tyrosine kinases (Janes, Nievergall & Lackmann Reference Janes, Nievergall and Lackmann2012; Case, Ditlev & Rosen Reference Case, Ditlev and Rosen2019) and simulation of membrane adhered to heterogenous substrate (Shelby et al. Reference Shelby, Castello-Serrano, Wisser, Levental and Veatch2023). Excess fluid from the adhesion patches is squeezed into the regions where molecules are unbound, which further increases the distance between membranes, creating pockets of fluids known as lumens, which can be observed in mammalian embryo development (Dumortier et al. Reference Dumortier, Le Verge-Serandour, Tortorelli, Mielke, de Plater, Turlier and Maître2019). These lumens can also be reproduced in reconstituted systems by applying an osmotic shock to a giant unilamellar vesicle (GUV) that is adhered to a supported lipid bilayer (Dinet et al. Reference Dinet, Torres-Sánchez, Lanfranco, Di Michele, Arroyo and Staykova2023). In these systems, the separated phases often undergo a passive, physically driven coarsening process where small patches diminish, while larger patches grow. These dynamics are reminiscent of other phase separation processes occurring in the cooling of metal alloys (Allen & Cahn Reference Allen and Cahn1979; Komura et al. Reference Komura, Osamura, Fujii and Takeda1985; Bray Reference Bray1994; Livet et al. Reference Livet, Bley, Caudron, Geissler, Abernathy, Detlefs, Grübel and Sutton2001), liquid–liquid phase separation in biological systems (Su et al. Reference Su, Ho, Gettel, Rowland, Keating and Parikh2024), as well as in droplet aggregation when a thin liquid or polymer film is adhered to a solid substrate by an attractive potential due to intermolecular forces (Derrida, Godrèche & Yekutieli Reference Derrida, Godrèche and Yekutieli1991; Otto, Rump & Slepcev Reference Otto, Rump and Slepcev2006; Gratton & Witelski Reference Gratton and Witelski2008; Lal et al. Reference Lal, Lurio, Liang, Narayanan, Darling and Sutton2020). In those systems, an effective interfacial tension drives the coarsening process, providing a well-established
 $t^{1/3}$
power law (Bray Reference Bray1994) for the growth the characteristic length scale
$t^{1/3}$
power law (Bray Reference Bray1994) for the growth the characteristic length scale
 $L_c$
. For the coarsening observed in biological membranes, while membrane tension could give rise to the same
$L_c$
. For the coarsening observed in biological membranes, while membrane tension could give rise to the same
 $1/3$
power law, the effect of membrane bending has not been studied and will potentially introduce different dynamics.
$1/3$
power law, the effect of membrane bending has not been studied and will potentially introduce different dynamics.
In this article, numerical simulations of lubrication flow including thermal fluctuations are used to study membrane adhesion, with particular emphasis on the effects of membrane bending as opposed to analogous systems involving interfacial tension. The average waiting time for adhesion to occur due to fluctuations is predicted using rare-event theory. We then show that membrane bending gives rise to power-law coarsening behaviour that is slower than for tension-driven coarsening. We also demonstrate that the strength of thermal fluctuations does not significantly alter the coarsening rate, but that the initial height of the membranes does so in the hydrodynamic regime.
2. Mathematical model and numerical methods
To simultaneously study the fluid flow, elastic membrane bending, protein-like binding and stochastic fluctuations during membrane adhesion, we turn to the elastohydrodynamic thin film (Williams & Davis Reference Williams and Davis1982; Carlson & Mahadevan Reference Carlson and Mahadevan2016), describing the physical scenario of figure 1
 $(a)$
, i.e. a thin layer of viscous liquid confined by an elastic membrane. For simplicity as well as reflecting in vitro experimental conditions in which a GUV interacts with a supported lipid bilayer (Fenz et al. Reference Fenz, Bihr, Schmidt, Merkel, Seifert, Sengupta and Smith2017; Dinet et al. Reference Dinet, Torres-Sánchez, Lanfranco, Di Michele, Arroyo and Staykova2023), the substrate is static while the upper membrane ‘rests’ on the viscous fluid film at height
$(a)$
, i.e. a thin layer of viscous liquid confined by an elastic membrane. For simplicity as well as reflecting in vitro experimental conditions in which a GUV interacts with a supported lipid bilayer (Fenz et al. Reference Fenz, Bihr, Schmidt, Merkel, Seifert, Sengupta and Smith2017; Dinet et al. Reference Dinet, Torres-Sánchez, Lanfranco, Di Michele, Arroyo and Staykova2023), the substrate is static while the upper membrane ‘rests’ on the viscous fluid film at height
 $\hat {h}(\hat {x},\hat {y},\hat {t})$
(where the hats indicate dimensional variables) and can move vertically. As in the biological and synthetic systems described previously (Dumortier et al. Reference Dumortier, Le Verge-Serandour, Tortorelli, Mielke, de Plater, Turlier and Maître2019; Dinet et al. Reference Dinet, Torres-Sánchez, Lanfranco, Di Michele, Arroyo and Staykova2023), the film height
$\hat {h}(\hat {x},\hat {y},\hat {t})$
(where the hats indicate dimensional variables) and can move vertically. As in the biological and synthetic systems described previously (Dumortier et al. Reference Dumortier, Le Verge-Serandour, Tortorelli, Mielke, de Plater, Turlier and Maître2019; Dinet et al. Reference Dinet, Torres-Sánchez, Lanfranco, Di Michele, Arroyo and Staykova2023), the film height
 $\hat {h}$
(which is typically in the range of tens of nanometres) is much smaller than the length of the domain in the lateral directions,
$\hat {h}$
(which is typically in the range of tens of nanometres) is much smaller than the length of the domain in the lateral directions,
 $L$
(typically several microns). Across the gap, membrane-bound proteins may bind or unbind to the solid surface.
$L$
(typically several microns). Across the gap, membrane-bound proteins may bind or unbind to the solid surface.

Figure 1.
 $(a)$
A sketch of an elastic membrane with thickness
$(a)$
A sketch of an elastic membrane with thickness
 $d$
in close proximity to a rigid wall separated by a thin layer of viscous fluid of height
$d$
in close proximity to a rigid wall separated by a thin layer of viscous fluid of height
 $\hat {h}(\hat {x},\hat {y},\hat {t})$
. Membrane molecules may bind across the channel only if their distance is below a critical value
$\hat {h}(\hat {x},\hat {y},\hat {t})$
. Membrane molecules may bind across the channel only if their distance is below a critical value
 $h^*$
.
$h^*$
.
 $(b)$
Left: a contour map of the non-dimensional height profile
$(b)$
Left: a contour map of the non-dimensional height profile
 $h(x,y,t)$
at the time
$h(x,y,t)$
at the time
 $t^*$
, which is the onset of adhesion between the membrane and solid surface. Data are shown for a membrane with non-dimensional initial height
$t^*$
, which is the onset of adhesion between the membrane and solid surface. Data are shown for a membrane with non-dimensional initial height
 $h_0=1.6$
and fluctuation intensity
$h_0=1.6$
and fluctuation intensity
 $Q_B=( {1}/{l})\sqrt {{2 k_B T }/{B^{1/2}(\kappa c_0)^{3/2}}}=0.01$
, with
$Q_B=( {1}/{l})\sqrt {{2 k_B T }/{B^{1/2}(\kappa c_0)^{3/2}}}=0.01$
, with
 $l$
the equilibrium length of the adhesive molecules,
$l$
the equilibrium length of the adhesive molecules,
 $k_BT$
the thermal energy,
$k_BT$
the thermal energy,
 $B$
the bending stiffness,
$B$
the bending stiffness,
 $c_0$
the equilibrium concentration and
$c_0$
the equilibrium concentration and
 $\kappa$
the molecule spring stiffness coefficient. Right: a cross-section of the profile along the red line in the contour map; the film height at the point of contact drops to a non-dimensional value of
$\kappa$
the molecule spring stiffness coefficient. Right: a cross-section of the profile along the red line in the contour map; the film height at the point of contact drops to a non-dimensional value of
 $h=\hat {h}/l\approx 1$
, matching the equilibrium length of the adhesive molecules.
$h=\hat {h}/l\approx 1$
, matching the equilibrium length of the adhesive molecules.
 $(c)$
Left: a contour map at a later time when most of the membrane is bound, but liquid is collected in unbound patches. Right: a cross-section of the profile along the red line in the contour map, illustrating the formation of blisters (regions where the adhesive molecules are unbound and
$(c)$
Left: a contour map at a later time when most of the membrane is bound, but liquid is collected in unbound patches. Right: a cross-section of the profile along the red line in the contour map, illustrating the formation of blisters (regions where the adhesive molecules are unbound and
 $h=\hat {h}/l\gtrapprox 1$
) during the coarsening process.
$h=\hat {h}/l\gtrapprox 1$
) during the coarsening process.
2.1. Stochastic thin film equation
By assuming a small aspect ratio of the viscous channel,
 $\hat {h}/L\ll 1$
, one can apply the lubrication approximation to describe the flow of a Newtonian fluid with viscosity
$\hat {h}/L\ll 1$
, one can apply the lubrication approximation to describe the flow of a Newtonian fluid with viscosity
 $\mu$
in the channel, leading to a parabolic velocity profile (Batchelor Reference Batchelor2000). A random stress tensor is introduced in the momentum equations to account for the thermal fluctuations in the fluid, which are significant on the relevant nanometric scale (Landau & Lifshitz Reference Landau and Lifshitz1987). By imposing no-slip and kinematic boundary conditions at the membrane, one arrives at the following stochastic thin film (Davidovitch, Moro & Stone Reference Davidovitch, Moro and Stone2005; Mecke & Rauscher Reference Mecke and Rauscher2005; Grün et al. Reference Grün, Mecke and Rauscher2006; Durán-Olivencia et al. Reference Durán-Olivencia, Gvalani, Kalliadasis and Pavliotis2019) that can be used to describe
$\mu$
in the channel, leading to a parabolic velocity profile (Batchelor Reference Batchelor2000). A random stress tensor is introduced in the momentum equations to account for the thermal fluctuations in the fluid, which are significant on the relevant nanometric scale (Landau & Lifshitz Reference Landau and Lifshitz1987). By imposing no-slip and kinematic boundary conditions at the membrane, one arrives at the following stochastic thin film (Davidovitch, Moro & Stone Reference Davidovitch, Moro and Stone2005; Mecke & Rauscher Reference Mecke and Rauscher2005; Grün et al. Reference Grün, Mecke and Rauscher2006; Durán-Olivencia et al. Reference Durán-Olivencia, Gvalani, Kalliadasis and Pavliotis2019) that can be used to describe
 $\hat {h}(\hat {x},\hat {y},\hat {t})$
:
$\hat {h}(\hat {x},\hat {y},\hat {t})$
:
 \begin{equation} \frac {\partial \hat {h}(\hat {x},\hat {y},\hat {t})}{\partial \hat {t}} = \hat {\boldsymbol{\nabla }}\boldsymbol{\cdot }\left (\frac {\hat {h}^3(\hat {x},\hat {y},\hat {t})}{12\mu }\hat {\boldsymbol{\nabla }} \hat {p}(\hat {x},\hat {y},\hat {t}) \right) + \sqrt {\frac {k_B T}{6\mu }}\hat {\boldsymbol{\nabla }} \boldsymbol{\cdot }\left (\hat {h}^{3/2}(\hat {x},\hat {y},\hat {t})\boldsymbol{\hat {\eta }}(\hat {x},\hat {y},\hat {t})\right) \end{equation}
\begin{equation} \frac {\partial \hat {h}(\hat {x},\hat {y},\hat {t})}{\partial \hat {t}} = \hat {\boldsymbol{\nabla }}\boldsymbol{\cdot }\left (\frac {\hat {h}^3(\hat {x},\hat {y},\hat {t})}{12\mu }\hat {\boldsymbol{\nabla }} \hat {p}(\hat {x},\hat {y},\hat {t}) \right) + \sqrt {\frac {k_B T}{6\mu }}\hat {\boldsymbol{\nabla }} \boldsymbol{\cdot }\left (\hat {h}^{3/2}(\hat {x},\hat {y},\hat {t})\boldsymbol{\hat {\eta }}(\hat {x},\hat {y},\hat {t})\right) \end{equation}
where
 $\hat {\boldsymbol{\nabla }}$
represents the two-dimensional (2-D) gradient operator
$\hat {\boldsymbol{\nabla }}$
represents the two-dimensional (2-D) gradient operator
 $(\partial /\partial \hat {x},\partial /\partial \hat {y})$
and the dependent variable
$(\partial /\partial \hat {x},\partial /\partial \hat {y})$
and the dependent variable
 $\hat h$
represents height in the third spatial direction.
$\hat h$
represents height in the third spatial direction.
The first term on the right-hand side of (2.1) represents the change in film height due to a lateral flux driven by the horizontal pressure gradient
 $\hat {\boldsymbol{\nabla }} \hat {p}$
. The second term on the right-hand side incorporates the effect of thermal fluctuations at temperature
$\hat {\boldsymbol{\nabla }} \hat {p}$
. The second term on the right-hand side incorporates the effect of thermal fluctuations at temperature
 $T$
, by introducing a random flux in accordance with the fluctuation–dissipation theorem and averaged across the channel (Davidovitch et al. Reference Davidovitch, Moro and Stone2005; Mecke & Rauscher Reference Mecke and Rauscher2005; Grün et al. Reference Grün, Mecke and Rauscher2006). Here,
$T$
, by introducing a random flux in accordance with the fluctuation–dissipation theorem and averaged across the channel (Davidovitch et al. Reference Davidovitch, Moro and Stone2005; Mecke & Rauscher Reference Mecke and Rauscher2005; Grün et al. Reference Grün, Mecke and Rauscher2006). Here,
 $\boldsymbol{\hat {\eta }} (\hat {x},\hat {y},\hat {t})$
, is a random vector with Gaussian white noise uncorrelated in both time and space, i.e.
$\boldsymbol{\hat {\eta }} (\hat {x},\hat {y},\hat {t})$
, is a random vector with Gaussian white noise uncorrelated in both time and space, i.e.
 $\langle \hat {\eta }_i(\hat {x},\hat {y},\hat {t})\rangle =0$
and
$\langle \hat {\eta }_i(\hat {x},\hat {y},\hat {t})\rangle =0$
and
 $\langle \hat {\eta }_i(\hat {x},\hat {y},\hat {t})\hat {\eta }_j(\hat {x}^{\prime},\hat {y}^{\prime},\hat {t}^{\prime})\rangle =\delta _{ij}\delta (\hat {x}-\hat {x}^{\prime})\delta (\hat {y}-\hat {y}^{\prime})\delta (\hat {t}-\hat {t}^{\prime})$
, where
$\langle \hat {\eta }_i(\hat {x},\hat {y},\hat {t})\hat {\eta }_j(\hat {x}^{\prime},\hat {y}^{\prime},\hat {t}^{\prime})\rangle =\delta _{ij}\delta (\hat {x}-\hat {x}^{\prime})\delta (\hat {y}-\hat {y}^{\prime})\delta (\hat {t}-\hat {t}^{\prime})$
, where
 $\langle \; \rangle$
is the ensemble average,
$\langle \; \rangle$
is the ensemble average,
 $\delta _{ij}$
is the Kronecker symbol,
$\delta _{ij}$
is the Kronecker symbol,
 $\delta$
is the Dirac distribution and
$\delta$
is the Dirac distribution and
 $k_B$
is the Boltzmann constant.
$k_B$
is the Boltzmann constant.
Here, we note that the nonlinear prefactor
 $\hat {h}^3/(12\mu)$
arises from viscous resistance to the flow, describing the mobility of the fluid. In fact, (2.1) can be re-written as a gradient flow (Durán-Olivencia et al. Reference Durán-Olivencia, Gvalani, Kalliadasis and Pavliotis2019; Cates Reference Cates2022; Sprittles et al. Reference Sprittles, Liu, Lockerby and Grafke2023) of the form
$\hat {h}^3/(12\mu)$
arises from viscous resistance to the flow, describing the mobility of the fluid. In fact, (2.1) can be re-written as a gradient flow (Durán-Olivencia et al. Reference Durán-Olivencia, Gvalani, Kalliadasis and Pavliotis2019; Cates Reference Cates2022; Sprittles et al. Reference Sprittles, Liu, Lockerby and Grafke2023) of the form
 \begin{equation} \frac {\partial \hat {h}(\hat {x},\hat {y},\hat {t})}{\partial \hat {t}} = \hat {\boldsymbol{\nabla }}\boldsymbol{\cdot }\left (M(\hat {h})\hat {\boldsymbol{\nabla }} \frac {\delta \hat {F}}{\delta \hat {h}} + \sqrt {2 k_B TM(\hat {h})}\boldsymbol{\hat {\eta }}\right)\!, \end{equation}
\begin{equation} \frac {\partial \hat {h}(\hat {x},\hat {y},\hat {t})}{\partial \hat {t}} = \hat {\boldsymbol{\nabla }}\boldsymbol{\cdot }\left (M(\hat {h})\hat {\boldsymbol{\nabla }} \frac {\delta \hat {F}}{\delta \hat {h}} + \sqrt {2 k_B TM(\hat {h})}\boldsymbol{\hat {\eta }}\right)\!, \end{equation}
where
 $M(\hat {h})$
is the mobility and
$M(\hat {h})$
is the mobility and
 $\hat {F}[\hat {h}(\hat {x},\hat {y})]$
is an energy functional that gives rise to the pressure. Depending on the problem and assumptions, the mobility can take other forms (Glasner Reference Glasner2008). For example, in a crowded, narrow section of cytoplasm, one could use Darcy’s law to describe the flow through a porous medium, which reduces the mobility to
$\hat {F}[\hat {h}(\hat {x},\hat {y})]$
is an energy functional that gives rise to the pressure. Depending on the problem and assumptions, the mobility can take other forms (Glasner Reference Glasner2008). For example, in a crowded, narrow section of cytoplasm, one could use Darcy’s law to describe the flow through a porous medium, which reduces the mobility to
 ${\sim} \hat {h}$
(Gnann & Petrache Reference Gnann and Petrache2018). A slip boundary, however, would enhance mobility by introducing an additional
${\sim} \hat {h}$
(Gnann & Petrache Reference Gnann and Petrache2018). A slip boundary, however, would enhance mobility by introducing an additional
 ${\sim} \hat {h}^2$
term (Zhang, Sprittles & Lockerby Reference Zhang, Sprittles and Lockerby2020). The
${\sim} \hat {h}^2$
term (Zhang, Sprittles & Lockerby Reference Zhang, Sprittles and Lockerby2020). The
 ${\sim} M^{1/2}$
prefactor of the fluctuation term ensures that detailed balance is satisfied (Durán-Olivencia et al. Reference Durán-Olivencia, Gvalani, Kalliadasis and Pavliotis2019; Liu et al. Reference Liu, Sprittles and Grafke2024). The free energy
${\sim} M^{1/2}$
prefactor of the fluctuation term ensures that detailed balance is satisfied (Durán-Olivencia et al. Reference Durán-Olivencia, Gvalani, Kalliadasis and Pavliotis2019; Liu et al. Reference Liu, Sprittles and Grafke2024). The free energy
 $\hat {F}$
depends on what forces drive the flux in the channel, as described in the following sections.
$\hat {F}$
depends on what forces drive the flux in the channel, as described in the following sections.
2.2. Membrane dynamics
Deformation of the membrane, as shown in figure 1
 $(a)$
, results in tension and bending forces, which lead to a change in the pressure
$(a)$
, results in tension and bending forces, which lead to a change in the pressure
 $\hat p(\hat {x},\hat {y},\hat {t})$
of the fluid in the channel. As the membrane changes shape, so does the fluid flux in accordance with (2.1). The membrane is idealised as an isotropic linearly elastic solid with Young’s modulus
$\hat p(\hat {x},\hat {y},\hat {t})$
of the fluid in the channel. As the membrane changes shape, so does the fluid flux in accordance with (2.1). The membrane is idealised as an isotropic linearly elastic solid with Young’s modulus
 $E$
, Poisson ratio
$E$
, Poisson ratio
 $\nu$
and thickness
$\nu$
and thickness
 $d$
(Landau & Lifshitz Reference Landau and Lifshitz1986). In the limit of small deflections, i.e. small spatial gradients in
$d$
(Landau & Lifshitz Reference Landau and Lifshitz1986). In the limit of small deflections, i.e. small spatial gradients in
 $\hat {h}(\hat {x},\hat {y},\hat {t})$
, which is natural from the scale separation, and small strains, the coupling between the pressure in the channel and the deflection of the membrane can be represented by the Föppl–von Kármán (FvK) equations (Audoly & Pomeau Reference Audoly and Pomeau2008; Howell, Kozyreff & Ockendon Reference Howell, Kozyreff and Ockendon2008). The FvK equations incorporate the effects of both bending and tension, the latter of which is highly coupled to the deformations in the membrane. We consider a membrane subject to constant tension
$\hat {h}(\hat {x},\hat {y},\hat {t})$
, which is natural from the scale separation, and small strains, the coupling between the pressure in the channel and the deflection of the membrane can be represented by the Föppl–von Kármán (FvK) equations (Audoly & Pomeau Reference Audoly and Pomeau2008; Howell, Kozyreff & Ockendon Reference Howell, Kozyreff and Ockendon2008). The FvK equations incorporate the effects of both bending and tension, the latter of which is highly coupled to the deformations in the membrane. We consider a membrane subject to constant tension
 $\gamma$
, which could arise from some external stretching of the membrane at its edges (or a pressure inside an adhered cell), which is much greater than the tension caused by local membrane deflections. Under this assumption, the relationship between membrane deflections and channel pressure becomes (Evans Reference Evans1985; Peng & Lister Reference Peng and Lister2019)
$\gamma$
, which could arise from some external stretching of the membrane at its edges (or a pressure inside an adhered cell), which is much greater than the tension caused by local membrane deflections. Under this assumption, the relationship between membrane deflections and channel pressure becomes (Evans Reference Evans1985; Peng & Lister Reference Peng and Lister2019)
 \begin{equation} \hat {p}_{\textit{elastic}}(\hat {x},\hat {y},\hat {t}) =B \hat {{\nabla }} ^4\hat {h}(\hat {x},\hat {y},\hat {t}) - \gamma \hat {{\nabla }} ^2\hat {h}(\hat {x},\hat {y},\hat {t}), \end{equation}
\begin{equation} \hat {p}_{\textit{elastic}}(\hat {x},\hat {y},\hat {t}) =B \hat {{\nabla }} ^4\hat {h}(\hat {x},\hat {y},\hat {t}) - \gamma \hat {{\nabla }} ^2\hat {h}(\hat {x},\hat {y},\hat {t}), \end{equation}
where
 $B=Ed^3/12(1-\nu ^2)$
is membrane bending rigidity and
$B=Ed^3/12(1-\nu ^2)$
is membrane bending rigidity and
 $\gamma$
is the membrane tension (which is analogous to the surface tension at a fluid–fluid interface). Both of these components are generally present, but their relative strength will depend on the ratio of the characteristic horizontal length scale of the system,
$\gamma$
is the membrane tension (which is analogous to the surface tension at a fluid–fluid interface). Both of these components are generally present, but their relative strength will depend on the ratio of the characteristic horizontal length scale of the system,
 $L$
, to the bendocapillary length
$L$
, to the bendocapillary length
 $l_{BC}= \sqrt {B/\gamma }$
(Deserno Reference Deserno2015). For
$l_{BC}= \sqrt {B/\gamma }$
(Deserno Reference Deserno2015). For
 $L/l_{BC}\ll 1$
, bending should be the prominent driving force, whereas for
$L/l_{BC}\ll 1$
, bending should be the prominent driving force, whereas for
 $L/l_{BC}\gg 1$
, we expect membrane tension to dominate. Note that the tension in a membrane can also be described by an integral constraint for the membrane length (Kodio, Griffiths & Vella Reference Kodio, Griffiths and Vella2017), or by solving the full FvK to give a more complete description of membrane deformation (Pihler-Puzović et al. Reference Pihler-Puzović, Périllat, Russell, Juel and Heil2013).
$L/l_{BC}\gg 1$
, we expect membrane tension to dominate. Note that the tension in a membrane can also be described by an integral constraint for the membrane length (Kodio, Griffiths & Vella Reference Kodio, Griffiths and Vella2017), or by solving the full FvK to give a more complete description of membrane deformation (Pihler-Puzović et al. Reference Pihler-Puzović, Périllat, Russell, Juel and Heil2013).
The pressure terms in (2.3) can be formulated as a gradient flow of (2.2) as the two contributions can be rewritten as functional derivatives of free energies. The bending energy is
 $\hat {F}_{{bend}}[\hat {h}(\hat {x},\hat {y})] = \int ({B}/{2}) |\hat \nabla^2 \hat h |^2\, {\rm d}\hat {A}$
and the surface energy is
$\hat {F}_{{bend}}[\hat {h}(\hat {x},\hat {y})] = \int ({B}/{2}) |\hat \nabla^2 \hat h |^2\, {\rm d}\hat {A}$
and the surface energy is
 $\hat {F}_{\gamma }[\hat {h}(\hat {x},\hat {y})] = \int ( {\gamma }/{2}) \lvert \hat {\boldsymbol{\nabla }} \hat {h}\rvert ^2 \,{\rm d}\hat {A}$
.
$\hat {F}_{\gamma }[\hat {h}(\hat {x},\hat {y})] = \int ( {\gamma }/{2}) \lvert \hat {\boldsymbol{\nabla }} \hat {h}\rvert ^2 \,{\rm d}\hat {A}$
.
2.3. Dynamics of adhesive molecules
When proteins or other adhesive molecules bind across the gap between the two membranes, additional forces are generated that contribute to the pressure
 $\hat p(\hat {x},\hat {y},\hat {t})$
(Carlson & Mahadevan Reference Carlson and Mahadevan2015a
,
Reference Carlson and Mahadevanb
). Here, we use a Hookean elastic spring model to describe how bound proteins resist stretching and compression. Given a concentration
$\hat p(\hat {x},\hat {y},\hat {t})$
(Carlson & Mahadevan Reference Carlson and Mahadevan2015a
,
Reference Carlson and Mahadevanb
). Here, we use a Hookean elastic spring model to describe how bound proteins resist stretching and compression. Given a concentration
 $\hat {c}(\hat {x},\hat {y},\hat {t})$
of the bound adhesive molecules, an additional contribution to the pressure
$\hat {c}(\hat {x},\hat {y},\hat {t})$
of the bound adhesive molecules, an additional contribution to the pressure
 $\hat p_{\textit{adh}}$
takes the form
$\hat p_{\textit{adh}}$
takes the form
 \begin{equation} \hat p_{\textit{adh}}(\hat {x},\hat {y},\hat {t})=\kappa (\hat {h}(\hat {x},\hat {y},\hat {t})-l)\hat {c}(\hat {x},\hat {y},\hat {t}), \end{equation}
\begin{equation} \hat p_{\textit{adh}}(\hat {x},\hat {y},\hat {t})=\kappa (\hat {h}(\hat {x},\hat {y},\hat {t})-l)\hat {c}(\hat {x},\hat {y},\hat {t}), \end{equation}
where
 $l$
is the equilibrium bond length and
$l$
is the equilibrium bond length and
 $\kappa$
is the spring coefficient.
$\kappa$
is the spring coefficient.
To determine
 $\hat {c}(\hat {x},\hat {y},\hat {t})$
, we use a minimal description of the chemical kinetics of a single protein species, a full description of this model is given in Appendix A (Bell, Dembo & Bongrand Reference Bell, Dembo and Bongrand1984; Carlson & Mahadevan Reference Carlson and Mahadevan2015a
). The adhesive molecules are assumed to be uniformly distributed on both surfaces at a constant concentration
$\hat {c}(\hat {x},\hat {y},\hat {t})$
, we use a minimal description of the chemical kinetics of a single protein species, a full description of this model is given in Appendix A (Bell, Dembo & Bongrand Reference Bell, Dembo and Bongrand1984; Carlson & Mahadevan Reference Carlson and Mahadevan2015a
). The adhesive molecules are assumed to be uniformly distributed on both surfaces at a constant concentration
 $c_0$
. The concentration of pairs of unbound proteins is thus
$c_0$
. The concentration of pairs of unbound proteins is thus
 $c_0-\hat {c}(\hat {x},\hat {y},\hat {t})$
. The molecules can bind or unbind at a rate of
$c_0-\hat {c}(\hat {x},\hat {y},\hat {t})$
. The molecules can bind or unbind at a rate of
 $(c_0-\hat c)K_{{on}}$
or
$(c_0-\hat c)K_{{on}}$
or
 $\hat cK_{\textit{off}}$
, respectively. The rate constant for binding,
$\hat cK_{\textit{off}}$
, respectively. The rate constant for binding,
 $K_{{on}}$
, is a Gaussian distribution about the equilibrium length of the bond,
$K_{{on}}$
, is a Gaussian distribution about the equilibrium length of the bond,
 $l$
, with a standard deviation
$l$
, with a standard deviation
 $\sigma$
; whereas the rate constant for unbinding,
$\sigma$
; whereas the rate constant for unbinding,
 $K_{\textit{off}}$
, is constant (Bell et al. Reference Bell, Dembo and Bongrand1984; Qi et al. Reference Qi, Groves and Chakraborty2001). Furthermore, we assume that the molecular dynamics of binding/unbinding are much faster than the time scale of viscosity-mediated membrane deformation, so that the adhesive molecule concentrations immediately adjust to the equilibrium values for a specific membrane height. Balancing the binding and unbinding rates then gives us the following expression for the concentration of a single species of bound molecules (Carlson & Mahadevan Reference Carlson and Mahadevan2015b
):
$K_{\textit{off}}$
, is constant (Bell et al. Reference Bell, Dembo and Bongrand1984; Qi et al. Reference Qi, Groves and Chakraborty2001). Furthermore, we assume that the molecular dynamics of binding/unbinding are much faster than the time scale of viscosity-mediated membrane deformation, so that the adhesive molecule concentrations immediately adjust to the equilibrium values for a specific membrane height. Balancing the binding and unbinding rates then gives us the following expression for the concentration of a single species of bound molecules (Carlson & Mahadevan Reference Carlson and Mahadevan2015b
):
 \begin{equation} \hat {c}(\hat {x},\hat {y},\hat {t})=c_0\frac {\exp \left (- ((\hat {h}(\hat {x},\hat {y},\hat {t})-l)/\sigma )^2\right)}{\exp \left (-((\hat {h}(\hat {x},\hat {y},\hat {t})-l)/\sigma)^2\right)+\tau _{{on}}/\tau _{\textit{off}}}, \end{equation}
\begin{equation} \hat {c}(\hat {x},\hat {y},\hat {t})=c_0\frac {\exp \left (- ((\hat {h}(\hat {x},\hat {y},\hat {t})-l)/\sigma )^2\right)}{\exp \left (-((\hat {h}(\hat {x},\hat {y},\hat {t})-l)/\sigma)^2\right)+\tau _{{on}}/\tau _{\textit{off}}}, \end{equation}
where a value of
 $\sigma /l=0.2$
is used in the results of this study and the ratio of kinetic times for binding to unbinding is set to
$\sigma /l=0.2$
is used in the results of this study and the ratio of kinetic times for binding to unbinding is set to
 $\tau _{on}/\tau _{\textit{off}}=1/3$
Carlson & Mahadevan (Reference Carlson and Mahadevan2015a
,Reference Carlson and Mahadevan
b
). We note that with
$\tau _{on}/\tau _{\textit{off}}=1/3$
Carlson & Mahadevan (Reference Carlson and Mahadevan2015a
,Reference Carlson and Mahadevan
b
). We note that with
 $\hat {c}(\hat {x},\hat {y},\hat {t})$
given by (2.5), the molecule kinetics can be reduced to a single function of
$\hat {c}(\hat {x},\hat {y},\hat {t})$
given by (2.5), the molecule kinetics can be reduced to a single function of
 $\hat {h}(\hat {x},\hat {y},\hat {t})$
, meaning that we do not need to solve for
$\hat {h}(\hat {x},\hat {y},\hat {t})$
, meaning that we do not need to solve for
 $\hat {c}(\hat {x},\hat {y},\hat {t})$
as a variable in our system, which would be the case if the time scale for molecules to bind or unbind were of the order of the time scale for viscous fluid transport in the channel. The pressure given by (2.4) and (2.5) can conveniently be written as a free energy
$\hat {c}(\hat {x},\hat {y},\hat {t})$
as a variable in our system, which would be the case if the time scale for molecules to bind or unbind were of the order of the time scale for viscous fluid transport in the channel. The pressure given by (2.4) and (2.5) can conveniently be written as a free energy
 $\hat {F}_{\textit{adh}}[h]$
, as is described in Appendix A.
$\hat {F}_{\textit{adh}}[h]$
, as is described in Appendix A.
2.4. Non-dimensional analysis
We work with non-dimensional versions of (2.1) in the remainder of this article. Different scalings of the lengths and time are used in accordance with the dominant physical effects for the work presented in the subsequent sections. In each case, (2.1) is left with one non-dimensional parameter,
 $Q$
, that is directly proportional to
$Q$
, that is directly proportional to
 $\langle |\delta \hat {h}|\rangle$
, the average amplitude of thermal fluctuations in the film (Dhaliwal et al. Reference Dhaliwal, Pedersen, Kadri, Miquelard-Garnier, Sollogoub, Peixinho, Salez and Carlson2024). Here, we outline the non-dimensionalisation used for each case; further details can be found in Appendix B.
$\langle |\delta \hat {h}|\rangle$
, the average amplitude of thermal fluctuations in the film (Dhaliwal et al. Reference Dhaliwal, Pedersen, Kadri, Miquelard-Garnier, Sollogoub, Peixinho, Salez and Carlson2024). Here, we outline the non-dimensionalisation used for each case; further details can be found in Appendix B.
In § 3, we study (2.1) on a one-dimensional (1-D) domain (which represents a 2-D film since
 $h$
is the height in the
$h$
is the height in the
 $z$
-direction), with
$z$
-direction), with
 $\gamma =0$
in (2.3) and no adhesive molecules/proteins. In this case, the horizontal length
$\gamma =0$
in (2.3) and no adhesive molecules/proteins. In this case, the horizontal length
 $\hat {x}$
is scaled by the domain size
$\hat {x}$
is scaled by the domain size
 $L$
, as it is the only relevant horizontal length scale, the film height
$L$
, as it is the only relevant horizontal length scale, the film height
 $\hat {h}(\hat {x},\hat {t})$
is scaled by the initial film height
$\hat {h}(\hat {x},\hat {t})$
is scaled by the initial film height
 $\hat {h}_0$
and time
$\hat {h}_0$
and time
 $\hat {t}$
is scaled by
$\hat {t}$
is scaled by
 $\tau _\mu = {12\mu L^6}/{B\hat {h}_0^3}$
, which is the time scale of viscous relaxation of an elastohydrodynamic thin film and can be obtained by scaling the left-hand side of (2.1) with the bending term on the right (Carlson Reference Carlson2018). With these scalings, the dimensionless version of (2.1) is
$\tau _\mu = {12\mu L^6}/{B\hat {h}_0^3}$
, which is the time scale of viscous relaxation of an elastohydrodynamic thin film and can be obtained by scaling the left-hand side of (2.1) with the bending term on the right (Carlson Reference Carlson2018). With these scalings, the dimensionless version of (2.1) is
 \begin{equation} \frac {\partial h}{\partial t} = \frac {\partial }{\partial x} \left (h^3\frac {\partial }{\partial x} \left (\frac {\partial ^4}{\partial x^4} h\right)\right) + Q_{1\text{-}\textrm{D}}\frac {\partial }{\partial x} \left (h^{3/2}\eta \right)\!, \end{equation}
\begin{equation} \frac {\partial h}{\partial t} = \frac {\partial }{\partial x} \left (h^3\frac {\partial }{\partial x} \left (\frac {\partial ^4}{\partial x^4} h\right)\right) + Q_{1\text{-}\textrm{D}}\frac {\partial }{\partial x} \left (h^{3/2}\eta \right)\!, \end{equation}
where
 $Q_{\textrm{1-D}}= ({L}/{\hat {h}_0})\sqrt { {2k_B T L}/{Bw}}$
. Here,
$Q_{\textrm{1-D}}= ({L}/{\hat {h}_0})\sqrt { {2k_B T L}/{Bw}}$
. Here,
 $w$
is the width of the quasi-one-dimensional (quasi-1-D) film in the y-direction.
$w$
is the width of the quasi-one-dimensional (quasi-1-D) film in the y-direction.
In § 4, we study (2.1) on a 2-D domain (i.e. a three-dimensional (3-D) film), with either
 $\gamma$
or
$\gamma$
or
 $B$
set to zero in (2.3) and the protein pressure contribution described in § 2.3. In this case, a physical horizontal length scale can be obtained by balancing the relevant membrane pressure with the protein spring pressure (Carlson & Mahadevan Reference Carlson and Mahadevan2015a
). When bending dominates, this gives
$B$
set to zero in (2.3) and the protein pressure contribution described in § 2.3. In this case, a physical horizontal length scale can be obtained by balancing the relevant membrane pressure with the protein spring pressure (Carlson & Mahadevan Reference Carlson and Mahadevan2015a
). When bending dominates, this gives
 $L_B= (B/c_0\kappa)^{1/4}$
and while tension dominates,
$L_B= (B/c_0\kappa)^{1/4}$
and while tension dominates,
 $L_\gamma =(\gamma /c_0\kappa)^{1/2}$
. We scale
$L_\gamma =(\gamma /c_0\kappa)^{1/2}$
. We scale
 $\hat {x}$
and
$\hat {x}$
and
 $\hat {y}$
by this length scale,
$\hat {y}$
by this length scale,
 $\hat {h}$
by the protein equilibrium length
$\hat {h}$
by the protein equilibrium length
 $l$
, the protein concentration
$l$
, the protein concentration
 $\hat {c}$
by
$\hat {c}$
by
 $c_0$
, and
$c_0$
, and
 $\hat {t}$
by a time scale constructed as for the 1-D case, but with the gradients scaled by the new horizontal length scale
$\hat {t}$
by a time scale constructed as for the 1-D case, but with the gradients scaled by the new horizontal length scale
 $L_\gamma$
or
$L_\gamma$
or
 $L_B$
. For the bending-driven case, inserting this scaling, i.e.
$L_B$
. For the bending-driven case, inserting this scaling, i.e.
 $\hat {x}=L_Bx$
,
$\hat {x}=L_Bx$
,
 $\hat {y}=L_By$
and
$\hat {y}=L_By$
and
 $\hat {t}=t( {12\mu B^{1/2}}/{l^3(c_0\kappa)^{3/2}})$
, gives the following dimensionless equation:
$\hat {t}=t( {12\mu B^{1/2}}/{l^3(c_0\kappa)^{3/2}})$
, gives the following dimensionless equation:
 \begin{equation} \text{Bending} \,(\gamma =0): \quad \frac {\partial h}{\partial t} = \boldsymbol{\nabla }\boldsymbol{\cdot }\left (h^3\boldsymbol{\nabla }\left ({\nabla} ^4h+ (h-1)c\right) \right) + Q_{B}\boldsymbol{\nabla }\boldsymbol{\cdot }\left (h^{3/2}\boldsymbol{\eta }\right)\!, \end{equation}
\begin{equation} \text{Bending} \,(\gamma =0): \quad \frac {\partial h}{\partial t} = \boldsymbol{\nabla }\boldsymbol{\cdot }\left (h^3\boldsymbol{\nabla }\left ({\nabla} ^4h+ (h-1)c\right) \right) + Q_{B}\boldsymbol{\nabla }\boldsymbol{\cdot }\left (h^{3/2}\boldsymbol{\eta }\right)\!, \end{equation}
where
 $Q_{B}= ({1}/{lL_B})\sqrt { {2k_B T }/{c_0\kappa }}$
. For tension-driven films, the equivalent scalings, i.e.
$Q_{B}= ({1}/{lL_B})\sqrt { {2k_B T }/{c_0\kappa }}$
. For tension-driven films, the equivalent scalings, i.e.
 $\hat {x}=L_\gamma x$
,
$\hat {x}=L_\gamma x$
,
 $\hat {y}=L_\gamma y$
and
$\hat {y}=L_\gamma y$
and
 $\hat {t}=t({12\mu \gamma }/{l^3(c_0\kappa)^{2}})$
, give us the appropriate form of the non-dimensional thin film equation:
$\hat {t}=t({12\mu \gamma }/{l^3(c_0\kappa)^{2}})$
, give us the appropriate form of the non-dimensional thin film equation:
 \begin{equation} \text{Tension}\, (B=0): \quad \frac {\partial h}{\partial t} = \boldsymbol{\nabla }\boldsymbol{\cdot }\left (h^3\boldsymbol{\nabla }\left (-{\nabla} ^2h+ (h-1)c\right) \right) + Q_{\gamma }\boldsymbol{\nabla }\boldsymbol{\cdot }\left (h^{3/2}\boldsymbol{\eta }\right)\!, \end{equation}
\begin{equation} \text{Tension}\, (B=0): \quad \frac {\partial h}{\partial t} = \boldsymbol{\nabla }\boldsymbol{\cdot }\left (h^3\boldsymbol{\nabla }\left (-{\nabla} ^2h+ (h-1)c\right) \right) + Q_{\gamma }\boldsymbol{\nabla }\boldsymbol{\cdot }\left (h^{3/2}\boldsymbol{\eta }\right)\!, \end{equation}
where
 $Q_{\gamma }=({1}/{lL_{\gamma }})\sqrt { {2k_B T }/{c_0\kappa }}$
.
$Q_{\gamma }=({1}/{lL_{\gamma }})\sqrt { {2k_B T }/{c_0\kappa }}$
.
2.5. Finite element solver
In this article, (2.6), (2.7) and (2.8) are solved using the finite element method on a rectangular domain. In all simulations, periodic boundary conditions are imposed in the horizontal directions at the four boundaries. Since the film height
 $h$
is a dependent variable, simulations are performed in both 1-D and 2-D domains, which represent 2-D and 3-D films, respectively. The source code for the simulations in this article is available on GitHub (Dhaliwal Reference Dhaliwal2025).
$h$
is a dependent variable, simulations are performed in both 1-D and 2-D domains, which represent 2-D and 3-D films, respectively. The source code for the simulations in this article is available on GitHub (Dhaliwal Reference Dhaliwal2025).
To solve (2.1), we set up a separate equation for the pressure
 $p$
, with contributions coming from (2.3) and (2.4). Equation (2.1) is divided into a system of two (plus an additional one for the film curvature
$p$
, with contributions coming from (2.3) and (2.4). Equation (2.1) is divided into a system of two (plus an additional one for the film curvature
 ${\nabla} ^2 h(x,y,t)$
when there is a bending pressure) coupled second-order partial differential equations. The system is reformulated into weak form where boundary terms can be neglected due to the periodic boundary conditions. The scalar fields
${\nabla} ^2 h(x,y,t)$
when there is a bending pressure) coupled second-order partial differential equations. The system is reformulated into weak form where boundary terms can be neglected due to the periodic boundary conditions. The scalar fields
 ${\nabla} ^2 h(x,y,t)$
,
${\nabla} ^2 h(x,y,t)$
,
 $p(x,y,t)$
and
$p(x,y,t)$
and
 $h(x,y,t)$
are then discretised with linear elements, and the system of equations is solved using a Newton’s method solver from the FEniCS finite element library (Logg et al. Reference Logg2012). Time integration is performed using an implicit first-order finite difference scheme. The domain and its discretisation in space and time are chosen according to the relevant non-dimensional form presented in § 2.4. For the 1-D simulations presented in § 3.2, a domain of length
$h(x,y,t)$
are then discretised with linear elements, and the system of equations is solved using a Newton’s method solver from the FEniCS finite element library (Logg et al. Reference Logg2012). Time integration is performed using an implicit first-order finite difference scheme. The domain and its discretisation in space and time are chosen according to the relevant non-dimensional form presented in § 2.4. For the 1-D simulations presented in § 3.2, a domain of length
 $L=1$
is used with
$L=1$
is used with
 $\Delta x=0.01$
and
$\Delta x=0.01$
and
 $\Delta t =1\times 10^{-8}$
. For the 2-D simulations presented in § 4.2, a square
$\Delta t =1\times 10^{-8}$
. For the 2-D simulations presented in § 4.2, a square
 $100\times 100$
-cell grid is used with both
$100\times 100$
-cell grid is used with both
 $\Delta x$
and
$\Delta x$
and
 $\Delta t$
varying in the range
$\Delta t$
varying in the range
 $1{-}3$
(which means that
$1{-}3$
(which means that
 $L$
is in the range
$L$
is in the range
 $100{-}300$
) for the various results presented.
$100{-}300$
) for the various results presented.
The random vector
 $\boldsymbol{\eta }(x,y,t)$
is implemented by choosing random numbers using the ‘normal’ function in the ‘random’ class of NUMPY (Harris et al. Reference Harris2020). For the numerical solution at each time step, the two components of
$\boldsymbol{\eta }(x,y,t)$
is implemented by choosing random numbers using the ‘normal’ function in the ‘random’ class of NUMPY (Harris et al. Reference Harris2020). For the numerical solution at each time step, the two components of
 $\boldsymbol{\eta }(x,y,t)$
are each assigned a new value at every point in the mesh. Values are drawn from a Gaussian distribution with zero mean and a variance of
$\boldsymbol{\eta }(x,y,t)$
are each assigned a new value at every point in the mesh. Values are drawn from a Gaussian distribution with zero mean and a variance of
 $1/(\Delta x \Delta t)$
in 1-D and
$1/(\Delta x \Delta t)$
in 1-D and
 $1/(\Delta x^2 \Delta t)$
in 2-D. Due to the stochastic nature of the problem, each individual run is not to be considered as deterministic. For each set of parameters, we run multiple independent realisations and report the ensemble averages of the extracted/predicted parameters.
$1/(\Delta x^2 \Delta t)$
in 2-D. Due to the stochastic nature of the problem, each individual run is not to be considered as deterministic. For each set of parameters, we run multiple independent realisations and report the ensemble averages of the extracted/predicted parameters.
3. Waiting time for the initiation of adhesion
Membrane adhesion is facilitated by the binding of membrane-anchored adhesive molecules, which requires them to be in close range. It is then natural to ask: how long does it take for fluctuations to deform the membranes sufficiently so that they can bind to initiate adhesion? In this section, we use the model described in § 2 to study the average waiting time for initial contact to occur. We limit the study to a 1-D periodic domain, to avoid excessive computational costs, but expect that our results should be qualitatively similar for a 2-D domain. The physical picture of our simplified model is shown in figure 1
 $(a)$
; the two membranes are separated by a channel of viscous fluid with initial uniform thickness
$(a)$
; the two membranes are separated by a channel of viscous fluid with initial uniform thickness
 $h_0$
. Although the channel height is initially only fluctuating about
$h_0$
. Although the channel height is initially only fluctuating about
 $h_0$
, some part of the domain eventually reaches a critical thickness
$h_0$
, some part of the domain eventually reaches a critical thickness
 $h^*$
at which the two adhesive molecules come in contact.
$h^*$
at which the two adhesive molecules come in contact.
3.1. Rare-event theory
It is well known that thermal fluctuations can generate waves across the membrane, where the average wave amplitudes
 $\delta h_q$
of frequency
$\delta h_q$
of frequency
 $q$
, or ‘roughness’ of the membrane, depends on bending moduli and tension coefficient (Helfrich & Servuss Reference Helfrich and Servuss1984; Rautu et al. Reference Rautu, Orsi, Michele, Rowlands, Cicuta and Turner2017). If the
$q$
, or ‘roughness’ of the membrane, depends on bending moduli and tension coefficient (Helfrich & Servuss Reference Helfrich and Servuss1984; Rautu et al. Reference Rautu, Orsi, Michele, Rowlands, Cicuta and Turner2017). If the
 $h_0-h^*$
is of the same order as
$h_0-h^*$
is of the same order as
 $\delta h_q$
, then thermal fluctuations can easily initiate binding. If
$\delta h_q$
, then thermal fluctuations can easily initiate binding. If
 $h_0-h^*$
is sufficiently larger than
$h_0-h^*$
is sufficiently larger than
 $\delta h_q$
, attachment of the molecules then requires the membrane profile
$\delta h_q$
, attachment of the molecules then requires the membrane profile
 $h(x,t)$
to attain a rather unlikely shape, which may take a long time. The process can thus be thought of as
$h(x,t)$
to attain a rather unlikely shape, which may take a long time. The process can thus be thought of as
 $h(x,y,t)$
fluctuating in an energy landscape that does not favour large deviations from
$h(x,y,t)$
fluctuating in an energy landscape that does not favour large deviations from
 $h_0$
, until it eventually gets ‘lucky’ and reaches
$h_0$
, until it eventually gets ‘lucky’ and reaches
 $h^*$
at some point in the domain. Although the waiting time for protein binding is a random variable, the rare event theory can provide a prediction for the ensemble-averaged waiting time, known as the Eyring–Kramers law, which states that
$h^*$
at some point in the domain. Although the waiting time for protein binding is a random variable, the rare event theory can provide a prediction for the ensemble-averaged waiting time, known as the Eyring–Kramers law, which states that
 $\langle t_B \rangle \sim C \exp ({2(F[h_B(x)]-F[h_0])/Q_{\text{1-D}}^2})$
, where
$\langle t_B \rangle \sim C \exp ({2(F[h_B(x)]-F[h_0])/Q_{\text{1-D}}^2})$
, where
 $\langle t_B \rangle$
is the ensemble-averaged binding time,
$\langle t_B \rangle$
is the ensemble-averaged binding time,
 $C$
is a prefactor,
$C$
is a prefactor,
 $F[h(x,t)]$
is the energy of a profile
$F[h(x,t)]$
is the energy of a profile
 $h(x,t)$
and
$h(x,t)$
and
 $h_B(x)$
is the final binding profile (Eyring Reference Eyring1935; Kramers Reference Kramers1940; Liu et al. Reference Liu, Sprittles and Grafke2024).
$h_B(x)$
is the final binding profile (Eyring Reference Eyring1935; Kramers Reference Kramers1940; Liu et al. Reference Liu, Sprittles and Grafke2024).
If the membrane primarily exhibits tension rather than bending, i.e.
 $L\gt l_{BC}$
, the energy is determined by the second term in (2.3), and the problem is analogous to the rupture of a thin viscous liquid film with a free surface due to van der Waals forces. This problem was recently studied by Sprittles et al. (Reference Sprittles, Liu, Lockerby and Grafke2023), where the rare-event description based on the Eyring–Kramers formula was validated by both numerical simulations of the stochastic thin film equations and in molecular dynamics simulations. In this paper, these methods are adapted to determining the binding time between two membranes when rather the bending dominates the pressure term in (2.3).
$L\gt l_{BC}$
, the energy is determined by the second term in (2.3), and the problem is analogous to the rupture of a thin viscous liquid film with a free surface due to van der Waals forces. This problem was recently studied by Sprittles et al. (Reference Sprittles, Liu, Lockerby and Grafke2023), where the rare-event description based on the Eyring–Kramers formula was validated by both numerical simulations of the stochastic thin film equations and in molecular dynamics simulations. In this paper, these methods are adapted to determining the binding time between two membranes when rather the bending dominates the pressure term in (2.3).
When bending dominates over tension, i.e.
 $L\lt l_{BC}$
, the non-dimensional energy functional for the 1-D membrane becomes
$L\lt l_{BC}$
, the non-dimensional energy functional for the 1-D membrane becomes
 \begin{equation} F[h(x)] = \int ^1_0 \frac {1}{2} \left (\frac {\partial ^2 h}{\partial x ^2}\right)^2 \,{\rm d}x. \end{equation}
\begin{equation} F[h(x)] = \int ^1_0 \frac {1}{2} \left (\frac {\partial ^2 h}{\partial x ^2}\right)^2 \,{\rm d}x. \end{equation}
The pressure term in (2.3) can be recovered by taking a functional derivative of
 $F$
with respect to
$F$
with respect to
 $h(x)$
. Finding the average waiting time for adhesion requires finding the profile
$h(x)$
. Finding the average waiting time for adhesion requires finding the profile
 $h_B(x)$
that minimises
$h_B(x)$
that minimises
 $F[h]$
while also maintaining conservation of mass, which enters as the constraint
$F[h]$
while also maintaining conservation of mass, which enters as the constraint
 $\int _0^1(h-h_0)\,{\rm d}x = 0$
, and satisfying the periodic boundary condition. This constrained optimisation problem can be solved using the Euler–Lagrange equation, which gives the fourth-order polynomial profile shown by the dashed blue line in figure 2
$\int _0^1(h-h_0)\,{\rm d}x = 0$
, and satisfying the periodic boundary condition. This constrained optimisation problem can be solved using the Euler–Lagrange equation, which gives the fourth-order polynomial profile shown by the dashed blue line in figure 2
 $(a)$
. The energy corresponding to this profile is
$(a)$
. The energy corresponding to this profile is
 $F_{B}= 360 (h_0-h^*)^2$
. With a sharp asymptotics analysis (see Appendix C for details), the predicted average attachment time is given by
$F_{B}= 360 (h_0-h^*)^2$
. With a sharp asymptotics analysis (see Appendix C for details), the predicted average attachment time is given by
 \begin{equation} \langle t_B \rangle = \frac {1}{\beta (h_B)}\sqrt {\frac {Q_{\text{1-D}}^2}{(2\pi)^7}} \exp \left (720\left (\frac {h_0-h^*}{Q_{\textrm{1-D}}}\right)^2\right)\!, \end{equation}
\begin{equation} \langle t_B \rangle = \frac {1}{\beta (h_B)}\sqrt {\frac {Q_{\text{1-D}}^2}{(2\pi)^7}} \exp \left (720\left (\frac {h_0-h^*}{Q_{\textrm{1-D}}}\right)^2\right)\!, \end{equation}
where the parameter
 $\beta$
is given by
$\beta$
is given by
 \begin{equation} \beta (h_B) = (h_0-h^*)(2\pi)^6\left (\frac {1}{2}h_0^3 +\frac {3}{8}h_0(h_0-h^*)^2\right)\!. \end{equation}
\begin{equation} \beta (h_B) = (h_0-h^*)(2\pi)^6\left (\frac {1}{2}h_0^3 +\frac {3}{8}h_0(h_0-h^*)^2\right)\!. \end{equation}
We note that the expressions in (3.2) and (3.3) predict the attachment time across the channel without any fitting parameters. They are valid in the limit of small noise, i.e. either large
 $h_0-h^*$
or small
$h_0-h^*$
or small
 $Q_{\text{1-D}}$
, such that the trajectory of initial attachment must cross
$Q_{\text{1-D}}$
, such that the trajectory of initial attachment must cross
 $h_B(x)$
in the energy landscape.
$h_B(x)$
in the energy landscape.

Figure 2.
 $(a)$
Film profiles at time of attachment obtained from 15 independent solutions (centred around the point of ‘contact’) with parameters
$(a)$
Film profiles at time of attachment obtained from 15 independent solutions (centred around the point of ‘contact’) with parameters
 $Q_{\text{1-D}}=5$
and
$Q_{\text{1-D}}=5$
and
 $h^*=0.3$
. The dotted black line represents the average of the individual simulations and the dashed blue line represents the theoretical prediction from the Euler–Lagrange equation.
$h^*=0.3$
. The dotted black line represents the average of the individual simulations and the dashed blue line represents the theoretical prediction from the Euler–Lagrange equation.
 $(b)$
Average waiting time for adhesion
$(b)$
Average waiting time for adhesion
 $\langle t^* \rangle$
as a function of
$\langle t^* \rangle$
as a function of
 $(h_0-h^*)^2$
for different values of the noise amplitude
$(h_0-h^*)^2$
for different values of the noise amplitude
 $Q_{\text{1-D}}$
. The lines represent the predicted value of
$Q_{\text{1-D}}$
. The lines represent the predicted value of
 $\langle t_B \rangle$
from (3.2) with no free parameter. Error bars represent the standard deviation for a set of
$\langle t_B \rangle$
from (3.2) with no free parameter. Error bars represent the standard deviation for a set of
 $N=15$
simulations for each data point. The shaded blue colour is intended as a guide to the eye to emphasise the region where rare-event theory is valid, i.e. attachment events are sufficiently unlikely.
$N=15$
simulations for each data point. The shaded blue colour is intended as a guide to the eye to emphasise the region where rare-event theory is valid, i.e. attachment events are sufficiently unlikely.
3.2. Numerical predictions
Numerical simulations of (2.6) are used to test the prediction in (3.2) considering only the effect of membrane bending. The simulations are initiated with a flat membrane of height
 $h_0=1$
. Since there are no adhesive molecules, i.e. no force pulling/pushing on the membrane, the minimum film height fluctuates randomly with time, occasionally reaching a lower value than it has before. The simulations are run until the lowest point in the film reaches the cutoff height
$h_0=1$
. Since there are no adhesive molecules, i.e. no force pulling/pushing on the membrane, the minimum film height fluctuates randomly with time, occasionally reaching a lower value than it has before. The simulations are run until the lowest point in the film reaches the cutoff height
 $h^*$
, indicating that the molecules are now within binding range for initial attachment, and we denote this time as
$h^*$
, indicating that the molecules are now within binding range for initial attachment, and we denote this time as
 $t^*$
. The point in the periodic domain at which attachment occurs is random. If the deformation required for film rupture,
$t^*$
. The point in the periodic domain at which attachment occurs is random. If the deformation required for film rupture,
 $h_0-h^*$
, is small, attachment occurs rapidly and the profile shape at
$h_0-h^*$
, is small, attachment occurs rapidly and the profile shape at
 $t^*$
varies significantly from simulation to simulation. When
$t^*$
varies significantly from simulation to simulation. When
 $h_0-h^*$
is larger, however, the minimum film height fluctuates for a long time before reaching attachment (the number of time steps being many orders of magnitude), often coming close many times before finally reaching
$h_0-h^*$
is larger, however, the minimum film height fluctuates for a long time before reaching attachment (the number of time steps being many orders of magnitude), often coming close many times before finally reaching
 $h^*$
. When this is the case, the individual profiles at
$h^*$
. When this is the case, the individual profiles at
 $t^*$
are consistent, as shown in figure 2
$t^*$
are consistent, as shown in figure 2
 $(a)$
, which suggests that attachment does indeed tend to occur at a specific minimum energy profile.
$(a)$
, which suggests that attachment does indeed tend to occur at a specific minimum energy profile.
When the value of
 $h_0-h^*$
is large, as is the case for the profiles shown in figure 2
$h_0-h^*$
is large, as is the case for the profiles shown in figure 2
 $(a)$
, one would expect that the final profile is similar to the fourth-order polynomial predicted theoretically in § 3.1. The dashed blue line in figure 2
$(a)$
, one would expect that the final profile is similar to the fourth-order polynomial predicted theoretically in § 3.1. The dashed blue line in figure 2
 $(a)$
shows that this is indeed the case if the profiles are centred about their minimum and then averaged. This indicates that it is truly rare for fluctuations to make the film reach
$(a)$
shows that this is indeed the case if the profiles are centred about their minimum and then averaged. This indicates that it is truly rare for fluctuations to make the film reach
 $h^*$
and that this almost always occurs very close to the minimum energy profile that allows attachment.
$h^*$
and that this almost always occurs very close to the minimum energy profile that allows attachment.
The average attachment time,
 $\langle t^* \rangle$
, is plotted for various values of
$\langle t^* \rangle$
, is plotted for various values of
 $Q_{\text{1-D}}$
and
$Q_{\text{1-D}}$
and
 $h^*$
in figure 2
$h^*$
in figure 2
 $(b)$
. It is clear that the time increases rapidly when
$(b)$
. It is clear that the time increases rapidly when
 $h^*$
is reduced and eventually grows exponentially with
$h^*$
is reduced and eventually grows exponentially with
 $(h_0-h^*)^2$
for constant
$(h_0-h^*)^2$
for constant
 $Q_{\text{1-D}}$
. The theoretical prediction for the attachment time across a membrane channel from (3.2) provides an excellent quantitative prediction for the average rupture time when
$Q_{\text{1-D}}$
. The theoretical prediction for the attachment time across a membrane channel from (3.2) provides an excellent quantitative prediction for the average rupture time when
 $h_0-h^*$
is large. For smaller values of
$h_0-h^*$
is large. For smaller values of
 $h_0-h^*$
, the rare-event theory is not valid, as the membrane profile does not necessarily cross the saddle point on its way to attachment. We note that the performance of the theoretical prediction also depends on
$h_0-h^*$
, the rare-event theory is not valid, as the membrane profile does not necessarily cross the saddle point on its way to attachment. We note that the performance of the theoretical prediction also depends on
 $Q_{\text{1-D}}$
, as fluctuations are more likely to cause large deformations to the film profile, making the event less rare as well as introducing larger errors in the Laplace method used to approximate the mean first passage time (see Appendix C). Generally, the rare-event prediction is valid when the attachment event is sufficiently rare, meaning that the average attachment time is sufficiently large. This can be achieved either by having a small value of
$Q_{\text{1-D}}$
, as fluctuations are more likely to cause large deformations to the film profile, making the event less rare as well as introducing larger errors in the Laplace method used to approximate the mean first passage time (see Appendix C). Generally, the rare-event prediction is valid when the attachment event is sufficiently rare, meaning that the average attachment time is sufficiently large. This can be achieved either by having a small value of
 $Q_{\text{1-D}}$
or a large height difference
$Q_{\text{1-D}}$
or a large height difference
 $h_0-h^*$
, as in the blue shaded region of figure 2
$h_0-h^*$
, as in the blue shaded region of figure 2
 $(b)$
.
$(b)$
.
4. Coarsening of adhesion patches
We now turn our attention to the next stage in the dynamics, when the adhesive molecules have formed bonds (
 $t \gt \langle t_B\rangle$
) that lead to a rich coarsening dynamics. To do this, we study (2.2) with the protein binding model of (2.4) and (2.5) on a 2-D domain, where we vary the membrane pressure term and the mobility factor.
$t \gt \langle t_B\rangle$
) that lead to a rich coarsening dynamics. To do this, we study (2.2) with the protein binding model of (2.4) and (2.5) on a 2-D domain, where we vary the membrane pressure term and the mobility factor.
Once adhesive molecules start to bind across the thin film, the membrane is pulled towards the substrate at the binding site, leading to further binding of adjacent proteins (Bell et al. Reference Bell, Dembo and Bongrand1984). This process brings the surfaces closer to each other, squeezing liquid out laterally through the channel. Conservation of the liquid, however, may lead to distinct regions where the membrane separation is small and proteins are bound or where membrane separation is large and proteins unbound, as is depicted in figure 1
 $(c)$
(Fenz et al. Reference Fenz, Bihr, Schmidt, Merkel, Seifert, Sengupta and Smith2017; Dinet et al. Reference Dinet, Torres-Sánchez, Lanfranco, Di Michele, Arroyo and Staykova2023). The network of unbound domains containing excess liquid may then coarsen to reduce the deformation of the membrane, i.e. reducing the overall energy of the system. Smaller pockets/lumen-like structures shrink as liquid is transported towards larger pockets/lumens, which subsequently grow in size. Such coarsening is reminiscent of when liquid droplets form in a dewetting liquid thin film (Otto et al. Reference Otto, Rump and Slepcev2006). It also falls into the broader category of coarsening dynamics in physical systems, which has been extensively studied both theoretically and experimentally (Hohenberg & Halperin Reference Hohenberg and Halperin1977; Furukawa Reference Furukawa1985; Bray Reference Bray1994; Camley & Brown Reference Camley and Brown2011; Cates Reference Cates2022). We will now describe how our model fits into this context, demonstrating that elastohydrodynamic thin films display distinct coarsening behaviour due to the combined effects of elasticity and viscosity.
$(c)$
(Fenz et al. Reference Fenz, Bihr, Schmidt, Merkel, Seifert, Sengupta and Smith2017; Dinet et al. Reference Dinet, Torres-Sánchez, Lanfranco, Di Michele, Arroyo and Staykova2023). The network of unbound domains containing excess liquid may then coarsen to reduce the deformation of the membrane, i.e. reducing the overall energy of the system. Smaller pockets/lumen-like structures shrink as liquid is transported towards larger pockets/lumens, which subsequently grow in size. Such coarsening is reminiscent of when liquid droplets form in a dewetting liquid thin film (Otto et al. Reference Otto, Rump and Slepcev2006). It also falls into the broader category of coarsening dynamics in physical systems, which has been extensively studied both theoretically and experimentally (Hohenberg & Halperin Reference Hohenberg and Halperin1977; Furukawa Reference Furukawa1985; Bray Reference Bray1994; Camley & Brown Reference Camley and Brown2011; Cates Reference Cates2022). We will now describe how our model fits into this context, demonstrating that elastohydrodynamic thin films display distinct coarsening behaviour due to the combined effects of elasticity and viscosity.
4.1. Domain coarsening of adhered patches
Equation (2.1) can be seen as the governing equation of a dynamical phase field, in which the film height
 $h(x,y,t)$
is the sole order parameter (McLeish et al. Reference McLeish, Cates, Higgins, Olmsted and Bray2003; Cates Reference Cates2022). When coupled with the pressure term given by (2.3), it is in fact quite similar to the widely studied dynamical model B, which is used to describe the coarsening of a conserved order parameter (Hohenberg & Halperin Reference Hohenberg and Halperin1977). Nevertheless, a number of features distinguish our system. First, a nonlinear
$h(x,y,t)$
is the sole order parameter (McLeish et al. Reference McLeish, Cates, Higgins, Olmsted and Bray2003; Cates Reference Cates2022). When coupled with the pressure term given by (2.3), it is in fact quite similar to the widely studied dynamical model B, which is used to describe the coarsening of a conserved order parameter (Hohenberg & Halperin Reference Hohenberg and Halperin1977). Nevertheless, a number of features distinguish our system. First, a nonlinear
 ${\sim} h^3$
mobility (as described in § 2.1) arises from the lubrication flow in the narrow channel. Second, rather than the typical double-well potential, our system imposes an energy landscape arising from the adhesive molecules/proteins described in § 2.3 and Appendix A. Finally, and perhaps most significantly, the classical Cahn–Hilliard Laplacian free energy term (which, in this case, represents isotropic membrane tension) is supplemented by the fourth-order bending term as described in (2.3).
${\sim} h^3$
mobility (as described in § 2.1) arises from the lubrication flow in the narrow channel. Second, rather than the typical double-well potential, our system imposes an energy landscape arising from the adhesive molecules/proteins described in § 2.3 and Appendix A. Finally, and perhaps most significantly, the classical Cahn–Hilliard Laplacian free energy term (which, in this case, represents isotropic membrane tension) is supplemented by the fourth-order bending term as described in (2.3).
A quantitative understanding of phase separating systems is achieved through the dynamical scaling hypothesis, which states that in the later stage of a coarsening process, the domain structure of a system (as quantified by the structure function
 $S(\boldsymbol {k},t)=\langle h_{\boldsymbol {k}}(t)h_{\boldsymbol {-k}}(t)\rangle$
) is constant in time if one rescales the lengths by a single characteristic length scale
$S(\boldsymbol {k},t)=\langle h_{\boldsymbol {k}}(t)h_{\boldsymbol {-k}}(t)\rangle$
) is constant in time if one rescales the lengths by a single characteristic length scale
 $L_c(t)$
(Bray Reference Bray1994; Camley & Brown Reference Camley and Brown2011). This concept has been shown to be valid both numerically and experimentally in many systems, with power-law growth displayed when
$L_c(t)$
(Bray Reference Bray1994; Camley & Brown Reference Camley and Brown2011). This concept has been shown to be valid both numerically and experimentally in many systems, with power-law growth displayed when
 $L_c(t)$
is computed as a moment of
$L_c(t)$
is computed as a moment of
 $S(\boldsymbol {k},t)$
or the radius of individual particles (Furukawa Reference Furukawa1985; Shinozaki & Oono Reference Shinozaki and Oono1993; Sung et al. Reference Sung, Karim, Douglas and Han1996; Tateno & Tanaka Reference Tateno and Tanaka2021; Saiseau et al. Reference Saiseau, Truong, Guérin, Delabre and Delville2024; Su et al. Reference Su, Ho, Gettel, Rowland, Keating and Parikh2024). For coarsening of the aforementioned model B system,
$S(\boldsymbol {k},t)$
or the radius of individual particles (Furukawa Reference Furukawa1985; Shinozaki & Oono Reference Shinozaki and Oono1993; Sung et al. Reference Sung, Karim, Douglas and Han1996; Tateno & Tanaka Reference Tateno and Tanaka2021; Saiseau et al. Reference Saiseau, Truong, Guérin, Delabre and Delville2024; Su et al. Reference Su, Ho, Gettel, Rowland, Keating and Parikh2024). For coarsening of the aforementioned model B system,
 $L_c(t)$
is known to grow according to a
$L_c(t)$
is known to grow according to a
 ${\sim} t^{1/3}$
power law, as has been validated by a number of numerical studies (Vladimirova, Malagoli & Mauri Reference Vladimirova, Malagoli and Mauri1998; Tiribocchi et al. Reference Tiribocchi, Wittkowski, Marenduzzo and Cates2015), and can also be predicted theoretically using scaling arguments, renormalisation group theory or Lifshitz–Slyozov–Wagner theory (in the limit where the minority phase occupies a small volume fraction) (Lifshitz & Slyozov Reference Lifshitz and Slyozov1961; Wagner Reference Wagner1961; Bray Reference Bray1994; Camley & Brown Reference Camley and Brown2011).
${\sim} t^{1/3}$
power law, as has been validated by a number of numerical studies (Vladimirova, Malagoli & Mauri Reference Vladimirova, Malagoli and Mauri1998; Tiribocchi et al. Reference Tiribocchi, Wittkowski, Marenduzzo and Cates2015), and can also be predicted theoretically using scaling arguments, renormalisation group theory or Lifshitz–Slyozov–Wagner theory (in the limit where the minority phase occupies a small volume fraction) (Lifshitz & Slyozov Reference Lifshitz and Slyozov1961; Wagner Reference Wagner1961; Bray Reference Bray1994; Camley & Brown Reference Camley and Brown2011).
For the case where the bending energy drives coarsening rather than surface tension, it is unclear if the coarsening rate will follow the
 ${\sim} t^{1/3}$
power law. This essentially corresponds to replacing the surface tension contribution to the classical free energy functional,
${\sim} t^{1/3}$
power law. This essentially corresponds to replacing the surface tension contribution to the classical free energy functional,
 $F_{\gamma }(h(x,y)) = \int ({1}/{2}) \lvert \boldsymbol{\nabla }h\rvert ^2 \,{\rm d}A$
, by the corresponding bending term
$F_{\gamma }(h(x,y)) = \int ({1}/{2}) \lvert \boldsymbol{\nabla }h\rvert ^2 \,{\rm d}A$
, by the corresponding bending term
 $F_{\textit{bend}}(h(x,y)) = \int ({1}/{2}) ({\nabla} ^2 h)^2 \,{\rm d}A$
. We now follow the derivation of Bray (Reference Bray1994) to make a scaling prediction for how bending changes the coarsening dynamics (details are provided in Appendix D.2). We ignore the effects of the nonlinear mobility as well as the specifics of the potential function. In analogy with the coarsening literature, we note that the pressure in our system takes a form that can be interpreted as a chemical potential
$F_{\textit{bend}}(h(x,y)) = \int ({1}/{2}) ({\nabla} ^2 h)^2 \,{\rm d}A$
. We now follow the derivation of Bray (Reference Bray1994) to make a scaling prediction for how bending changes the coarsening dynamics (details are provided in Appendix D.2). We ignore the effects of the nonlinear mobility as well as the specifics of the potential function. In analogy with the coarsening literature, we note that the pressure in our system takes a form that can be interpreted as a chemical potential
 $\varPhi$
(Hohenberg & Halperin Reference Hohenberg and Halperin1977; Bray Reference Bray1994; Cates Reference Cates2022). We thus adopt the notation
$\varPhi$
(Hohenberg & Halperin Reference Hohenberg and Halperin1977; Bray Reference Bray1994; Cates Reference Cates2022). We thus adopt the notation
 $ p\equiv \varPhi =\delta F/\delta h$
, where
$ p\equiv \varPhi =\delta F/\delta h$
, where
 $F$
consists of the aforementioned bending term and a symmetric double-well potential function
$F$
consists of the aforementioned bending term and a symmetric double-well potential function
 $V(h)$
. The chemical potential is thus
$V(h)$
. The chemical potential is thus
 \begin{equation} \varPhi = \frac {\partial V}{\partial h} +{\nabla} ^4h \end{equation}
\begin{equation} \varPhi = \frac {\partial V}{\partial h} +{\nabla} ^4h \end{equation}
and the film height then evolves according to a continuity equation with flux
 $\boldsymbol{j}=-\boldsymbol{\nabla }\varPhi$
.
$\boldsymbol{j}=-\boldsymbol{\nabla }\varPhi$
.
In late-stage coarsening when motion of the interface between two domains is slow, diffusion of the order parameter
 $h$
and chemical potential
$h$
and chemical potential
 $\varPhi$
in the bulk is fast, so both should satisfy Laplace’s equation
$\varPhi$
in the bulk is fast, so both should satisfy Laplace’s equation
 ${\nabla} ^2h={\nabla} ^2\varPhi =0$
in the bulk regions. The flux through the interface, and thus by continuity the velocity of the interface, can then be determined by finding
${\nabla} ^2h={\nabla} ^2\varPhi =0$
in the bulk regions. The flux through the interface, and thus by continuity the velocity of the interface, can then be determined by finding
 $\varPhi$
at the interface.
$\varPhi$
at the interface.
At an interface between the two phases with radius of curvature
 $R$
,
$R$
,
 $\varPhi$
can be re-expressed in terms of the coordinate
$\varPhi$
can be re-expressed in terms of the coordinate
 $g$
representing distance along the unit vector
$g$
representing distance along the unit vector
 $\boldsymbol{\hat {g}}$
in the direction perpendicular to the interface (
$\boldsymbol{\hat {g}}$
in the direction perpendicular to the interface (
 $g=\pm \infty$
in the bulk and
$g=\pm \infty$
in the bulk and
 $g=0$
at the centre of the interface). Noting that
$g=0$
at the centre of the interface). Noting that
 $\boldsymbol{\nabla }h = (\partial h/\partial g)\boldsymbol{\hat {g}}$
and
$\boldsymbol{\nabla }h = (\partial h/\partial g)\boldsymbol{\hat {g}}$
and
 $\boldsymbol{\nabla }\boldsymbol{\cdot }\boldsymbol{\hat {g}}=1/R$
near the interface, we find that
$\boldsymbol{\nabla }\boldsymbol{\cdot }\boldsymbol{\hat {g}}=1/R$
near the interface, we find that
 \begin{equation} \varPhi = \frac {\partial V}{\partial h} +\frac {\partial ^4 h}{\partial g^4}+\frac {2}{R}\frac {\partial ^3 h}{\partial g^3}-\frac {1}{R^2}\frac {\partial ^2 h}{\partial g^2}+\frac {1}{R^3}\frac {\partial h}{\partial g}. \end{equation}
\begin{equation} \varPhi = \frac {\partial V}{\partial h} +\frac {\partial ^4 h}{\partial g^4}+\frac {2}{R}\frac {\partial ^3 h}{\partial g^3}-\frac {1}{R^2}\frac {\partial ^2 h}{\partial g^2}+\frac {1}{R^3}\frac {\partial h}{\partial g}. \end{equation}
The value of
 $\varPhi$
at the interface is then determined by multiplying (4.2) by
$\varPhi$
at the interface is then determined by multiplying (4.2) by
 $\partial h/\partial g$
and integrating across the interface from
$\partial h/\partial g$
and integrating across the interface from
 $g=-\infty$
to
$g=-\infty$
to
 $g=\infty$
. By imposing the boundary condition that
$g=\infty$
. By imposing the boundary condition that
 $h$
is constant in the bulk and assuming that the chemical potential is a double well with equal potential on both sides, integration gives
$h$
is constant in the bulk and assuming that the chemical potential is a double well with equal potential on both sides, integration gives
 \begin{equation} \varPhi \Delta h = -\frac {2}{R}\int ^\infty _{-\infty }\left (\frac {\partial ^2 h}{\partial g^2}\right)^2\,{\rm d}g+\frac {1}{R^3}\int ^\infty _{-\infty }\left (\frac {\partial h}{\partial g}\right)^2\,{\rm d}g, \end{equation}
\begin{equation} \varPhi \Delta h = -\frac {2}{R}\int ^\infty _{-\infty }\left (\frac {\partial ^2 h}{\partial g^2}\right)^2\,{\rm d}g+\frac {1}{R^3}\int ^\infty _{-\infty }\left (\frac {\partial h}{\partial g}\right)^2\,{\rm d}g, \end{equation}
where
 $\Delta h$
is the height difference between the domains on the inside and outside of the interface, and the integrals
$\Delta h$
is the height difference between the domains on the inside and outside of the interface, and the integrals
 $-2\int ^\infty _{-\infty} ({\partial ^2 h}/{\partial g^2})^2\,{\rm d}g$
and
$-2\int ^\infty _{-\infty} ({\partial ^2 h}/{\partial g^2})^2\,{\rm d}g$
and
 $\int ^\infty _{-\infty} ({\partial h}/{\partial g})^2\,{\rm d}g$
can effectively be interpreted as 2-D line tension-like coefficients
$\int ^\infty _{-\infty} ({\partial h}/{\partial g})^2\,{\rm d}g$
can effectively be interpreted as 2-D line tension-like coefficients
 $\varGamma _1$
and
$\varGamma _1$
and
 $\varGamma _2$
that represent an energy per unit length associated with the existence of the interface between the two phases in 2-D space. Equation (4.3) thus represents a Gibbs–Thomson-like boundary condition that determines the value of
$\varGamma _2$
that represent an energy per unit length associated with the existence of the interface between the two phases in 2-D space. Equation (4.3) thus represents a Gibbs–Thomson-like boundary condition that determines the value of
 $\varPhi$
on interfaces with radius of curvature
$\varPhi$
on interfaces with radius of curvature
 $R$
. Since the flux of fluid is proportional to the gradient of the chemical potential (assuming a constant mobility), the velocity of the interface scales as
$R$
. Since the flux of fluid is proportional to the gradient of the chemical potential (assuming a constant mobility), the velocity of the interface scales as
 $-\partial \varPhi /\partial g$
. Assuming that the domains are circular with radius
$-\partial \varPhi /\partial g$
. Assuming that the domains are circular with radius
 $R$
corresponding to the macroscopic length scale
$R$
corresponding to the macroscopic length scale
 $L_c$
, this then gives the following growth law for bending-driven coarsening:
$L_c$
, this then gives the following growth law for bending-driven coarsening:
 \begin{equation} \frac {{\rm d}L_c}{{\rm d}t}\sim \frac {\varGamma _1}{L_c^2}+\frac {\varGamma _2}{L_c^4}. \end{equation}
\begin{equation} \frac {{\rm d}L_c}{{\rm d}t}\sim \frac {\varGamma _1}{L_c^2}+\frac {\varGamma _2}{L_c^4}. \end{equation}
Equation (4.4) stands in contrast to the model B case which has only a single contribution to the line tension, giving rise to an
 ${\sim} L_c^{-2}$
growth rate. This suggests that bending-driven coarsening should have a coarsening rate with an exponent somewhere between
${\sim} L_c^{-2}$
growth rate. This suggests that bending-driven coarsening should have a coarsening rate with an exponent somewhere between
 $1/3$
and
$1/3$
and
 $1/5$
with time, with the value being determined by the relative sizes of
$1/5$
with time, with the value being determined by the relative sizes of
 $\varGamma _1$
and
$\varGamma _1$
and
 $\varGamma _2$
, which in turn depend on the shape of the potential function. We note that this simple scaling prediction only describes the difference between surface tension-driven and bending-driven coarsening. It does not take into account the other differences between our model and the classical model B system described by Bray (Reference Bray1994) such as the
$\varGamma _2$
, which in turn depend on the shape of the potential function. We note that this simple scaling prediction only describes the difference between surface tension-driven and bending-driven coarsening. It does not take into account the other differences between our model and the classical model B system described by Bray (Reference Bray1994) such as the
 ${\sim} h^3$
mobility and the protein binding potential energy. Since the potential energy function
${\sim} h^3$
mobility and the protein binding potential energy. Since the potential energy function
 $V(h)$
is not specified, we expect these results to be valid for other adhesive potentials such as van der Waals interactions. Equation (4.4) suggests a fundamental distinction between surface tension-driven and bending-driven coarsening, which will be investigated numerically for our specific system in § 4.2.
$V(h)$
is not specified, we expect these results to be valid for other adhesive potentials such as van der Waals interactions. Equation (4.4) suggests a fundamental distinction between surface tension-driven and bending-driven coarsening, which will be investigated numerically for our specific system in § 4.2.
4.2. Numerical investigation of bending-driven coarsening dynamics
We now present the results of numerical simulations of the phase separation of adhesive molecules/proteins during membrane adhesion when dominated by bending of the membrane, as described in (2.7). The membrane is always initialised as a flat profile with non-dimensional initial height
 $h(x,y,0)= \hat {h}_0/l\gt 1$
and the non-dimensional width of the protein binding rate distribution,
$h(x,y,0)= \hat {h}_0/l\gt 1$
and the non-dimensional width of the protein binding rate distribution,
 $\sigma _{{on}}/l=0.2$
. Figure 3(
$\sigma _{{on}}/l=0.2$
. Figure 3(
 $a$
) shows the contour plots (see supplementary movie 1 available at https://doi.org/10.1017/jfm.2025.10872) of the film profile at three different times when the initial film height is just at the edge of the range in which the proteins can bind. Fluctuations are clearly present for the first time steps, where the film is pulled to the substrate by the adhesive molecules. Most of the film profile moves towards the equilibrium length of the adhesive molecules (
$a$
) shows the contour plots (see supplementary movie 1 available at https://doi.org/10.1017/jfm.2025.10872) of the film profile at three different times when the initial film height is just at the edge of the range in which the proteins can bind. Fluctuations are clearly present for the first time steps, where the film is pulled to the substrate by the adhesive molecules. Most of the film profile moves towards the equilibrium length of the adhesive molecules (
 $h=1$
in non-dimensional terms) to reduce the energy of the system. Due to conservation of liquid mass, excess liquid must flow somewhere else, but periodic boundary conditions prevent fluid from leaving the domain. Thus, in some regions, the film is pushed up, forming circular ‘pockets’ or ‘lumens’ in which proteins are unbound and the film height is significantly larger than the initial height, as can be seen in the first panel to the left in figure 3(
$h=1$
in non-dimensional terms) to reduce the energy of the system. Due to conservation of liquid mass, excess liquid must flow somewhere else, but periodic boundary conditions prevent fluid from leaving the domain. Thus, in some regions, the film is pushed up, forming circular ‘pockets’ or ‘lumens’ in which proteins are unbound and the film height is significantly larger than the initial height, as can be seen in the first panel to the left in figure 3(
 $a$
).
$a$
).

Figure 3. Contour plots illustrating the height
 $h(x,y,t)$
of thin films in a 2-D domain under the influence of protein binding and unbinding for different times
$h(x,y,t)$
of thin films in a 2-D domain under the influence of protein binding and unbinding for different times
 $t$
, with
$t$
, with
 $h_0=1.4$
. In panel
$h_0=1.4$
. In panel
 $(a)$
, the fluctuation parameter is
$(a)$
, the fluctuation parameter is
 $Q_{B}=0.005$
, whereas in panel
$Q_{B}=0.005$
, whereas in panel
 $(b)$
, it is set to
$(b)$
, it is set to
 $Q_{B}=0.5$
. Although similar coarsening dynamics occur at late times regardless of
$Q_{B}=0.5$
. Although similar coarsening dynamics occur at late times regardless of
 $Q_{B}$
, the size of the domains at
$Q_{B}$
, the size of the domains at
 $t=350$
is somewhat larger for
$t=350$
is somewhat larger for
 $Q_{B}=0.5$
due to early-time coalescence.
$Q_{B}=0.5$
due to early-time coalescence.
With time, the detached domains coarsen in an Ostwald ripening-like process, resulting in fewer, larger domains as can be seen in the second and third panels of figure 3(
 $a$
). Once the circular pockets form, their centres remain essentially fixed as fluid is slowly transported from smaller to larger domains in a diffusion-like manner. When the size of a single pocket drops below a critical threshold, it suddenly ‘implodes’, which causes a minor horizontal rearrangement of nearby domains (see supplementary movies). Figure 4(
$a$
). Once the circular pockets form, their centres remain essentially fixed as fluid is slowly transported from smaller to larger domains in a diffusion-like manner. When the size of a single pocket drops below a critical threshold, it suddenly ‘implodes’, which causes a minor horizontal rearrangement of nearby domains (see supplementary movies). Figure 4(
 $a$
) shows the time evolution of the characteristic size
$a$
) shows the time evolution of the characteristic size
 $L_c$
for these domains, as computed using the method of Shinozaki & Oono (Reference Shinozaki and Oono1993), averaging the structure factor
$L_c$
for these domains, as computed using the method of Shinozaki & Oono (Reference Shinozaki and Oono1993), averaging the structure factor
 $S(\boldsymbol {k},t)$
across 15 independent realisations (details of how we calculte
$S(\boldsymbol {k},t)$
across 15 independent realisations (details of how we calculte
 $L_c$
are provided in Appendix D.1). Although the distinct ‘implosion’ events lead to discrete jumps in the growth for an individual simulation, the ensemble-averaged
$L_c$
are provided in Appendix D.1). Although the distinct ‘implosion’ events lead to discrete jumps in the growth for an individual simulation, the ensemble-averaged
 $L_c$
grows smoothly with time following a power law. The growth is significantly slower than the
$L_c$
grows smoothly with time following a power law. The growth is significantly slower than the
 ${\sim} t^{1/3}$
power law observed for a model B system (Bray Reference Bray1994; Tiribocchi et al. Reference Tiribocchi, Wittkowski, Marenduzzo and Cates2015), instead, the exponent is closer to
${\sim} t^{1/3}$
power law observed for a model B system (Bray Reference Bray1994; Tiribocchi et al. Reference Tiribocchi, Wittkowski, Marenduzzo and Cates2015), instead, the exponent is closer to
 $1/5$
, which can be expected based on our analysis in § 4.1.
$1/5$
, which can be expected based on our analysis in § 4.1.

Figure 4. Length scale
 $L_c$
computed using (D5) from the average of
$L_c$
computed using (D5) from the average of
 $N=15$
individual simulations under the same conditions as the data in figure 3. In panel
$N=15$
individual simulations under the same conditions as the data in figure 3. In panel
 $(a)$
, the fluctuation parameter is
$(a)$
, the fluctuation parameter is
 $Q_{B}=0.005$
, whereas in panel
$Q_{B}=0.005$
, whereas in panel
 $(b)$
, it is set to
$(b)$
, it is set to
 $Q_{B}=0.5$
. Power law coarsening with an exponent well below
$Q_{B}=0.5$
. Power law coarsening with an exponent well below
 $1/3$
is observed in both cases, but starts off with a larger domain size when
$1/3$
is observed in both cases, but starts off with a larger domain size when
 $Q_{B}=0.5$
.
$Q_{B}=0.5$
.
The contour plots in figure 3(
 $b$
) show the evolution of height profiles of a coarsening film when the amplitude of the thermal fluctuations, represented by the parameter
$b$
) show the evolution of height profiles of a coarsening film when the amplitude of the thermal fluctuations, represented by the parameter
 $Q_{B}$
, is increased by two orders of magnitude. With higher noise amplitude, the lumens are irregular in shape and constantly deforming (see supplementary movie 2). The fluctuations also cause significant lateral motion of the domains, which leads to some instances of coalescence at very early times. Despite this, at the later time, large droplets seem to repel each other and domain growth is primarily caused by fluid flow through adhered patches, as was observed in the low noise amplitude case. Interestingly, the power law for domain growth is relatively unaffected by the strength of the fluctuations, as shown in figure 4(
$Q_{B}$
, is increased by two orders of magnitude. With higher noise amplitude, the lumens are irregular in shape and constantly deforming (see supplementary movie 2). The fluctuations also cause significant lateral motion of the domains, which leads to some instances of coalescence at very early times. Despite this, at the later time, large droplets seem to repel each other and domain growth is primarily caused by fluid flow through adhered patches, as was observed in the low noise amplitude case. Interestingly, the power law for domain growth is relatively unaffected by the strength of the fluctuations, as shown in figure 4(
 $b$
). The size of the domains before the late-stage power law growth is somewhat higher when the fluctuations are larger, perhaps due to coalescence events at early times, as can be seen in the first panels of figure 3(a,b). The primary effect of fluctuations is thus to provide the perturbations that trigger the instabilities giving rise to the initial configuration, rather than driving coarsening.
$b$
). The size of the domains before the late-stage power law growth is somewhat higher when the fluctuations are larger, perhaps due to coalescence events at early times, as can be seen in the first panels of figure 3(a,b). The primary effect of fluctuations is thus to provide the perturbations that trigger the instabilities giving rise to the initial configuration, rather than driving coarsening.
To better understand how and why the coarsening rate of our system differs from the more familiar model B system, we perform additional simulations in which we vary the initial membrane height
 $h_0$
, the fluctuation strength
$h_0$
, the fluctuation strength
 $Q$
, the mobility prefactor in the thin film equation and which term in (2.3) we use. In general, we find that coarsening behaviour is a preserved feature of the system even when these parameters are changed. To ensure an initial configuration with many small pockets in the domain,
$Q$
, the mobility prefactor in the thin film equation and which term in (2.3) we use. In general, we find that coarsening behaviour is a preserved feature of the system even when these parameters are changed. To ensure an initial configuration with many small pockets in the domain,
 $h_0$
should be close to the edge of the binding range of the adhesive molecules. If
$h_0$
should be close to the edge of the binding range of the adhesive molecules. If
 $h_0$
is too small, the entire domain is already in a bound state and few pockets form. If, however,
$h_0$
is too small, the entire domain is already in a bound state and few pockets form. If, however,
 $h_0$
is too large, it takes a very long time for contact to occur, which is typically at only one point in the domain, leading to a small number of initial pockets when coarsening starts.
$h_0$
is too large, it takes a very long time for contact to occur, which is typically at only one point in the domain, leading to a small number of initial pockets when coarsening starts.
To begin with, we simulate coarsening using the tension term of (2.3) (corresponding to the well-studied Cahn–Hilliard free energy) instead of the bending term, as in (2.8). As expected, coarsening behaviour is observed, as shown in figure 5. Comparing the first snapshots of figures 5
 $(a)$
and 5
$(a)$
and 5
 $(b)$
demonstrates that larger initial heights lead to fewer domains at early times. At later times, we observe that the coarsening actually appears to be slower when
$(b)$
demonstrates that larger initial heights lead to fewer domains at early times. At later times, we observe that the coarsening actually appears to be slower when
 $h_0$
is increased.
$h_0$
is increased.

Figure 5. Contour plots illustrating the height
 $h(x,y,t)$
of thin films in a 2-D domain under the influence of protein binding and unbinding for different times
$h(x,y,t)$
of thin films in a 2-D domain under the influence of protein binding and unbinding for different times
 $t$
, with
$t$
, with
 $Q_{\gamma }=0.01$
. In panel
$Q_{\gamma }=0.01$
. In panel
 $(a)$
, the initial height is
$(a)$
, the initial height is
 $h_0=1.25$
, whereas in panel
$h_0=1.25$
, whereas in panel
 $(b)$
, it is set to
$(b)$
, it is set to
 $h_0=1.45$
. Increasing
$h_0=1.45$
. Increasing
 $h_0$
leads to a larger initial domain size, but slower coarsening at late times.
$h_0$
leads to a larger initial domain size, but slower coarsening at late times.
Figure 6
 $(a)$
shows how the characteristic length scale
$(a)$
shows how the characteristic length scale
 $L_c$
of a viscous film profile grows with time when the membrane exhibits only interfacial tension. When
$L_c$
of a viscous film profile grows with time when the membrane exhibits only interfacial tension. When
 $h_0$
is small, the growth exponent is close to
$h_0$
is small, the growth exponent is close to
 ${\sim} t^{1/3}$
, in accordance with what one expects for the well-known model B system. For higher values of
${\sim} t^{1/3}$
, in accordance with what one expects for the well-known model B system. For higher values of
 $h_0$
, the growth rate for late-stage coarsening decreases. This seems to be caused by the nonlinear
$h_0$
, the growth rate for late-stage coarsening decreases. This seems to be caused by the nonlinear
 ${\sim} h^3$
term arising from the viscous resistance to flow in the governing equation. To confirm this, we perform additional simulations in which we replace the nonlinear mobility by a constant mobility with value
${\sim} h^3$
term arising from the viscous resistance to flow in the governing equation. To confirm this, we perform additional simulations in which we replace the nonlinear mobility by a constant mobility with value
 $1$
. The inset in figure 6
$1$
. The inset in figure 6
 $(a)$
shows that
$(a)$
shows that
 $L_c\sim t^{1/3}$
growth is observed regardless of
$L_c\sim t^{1/3}$
growth is observed regardless of
 $h_0$
when the mobility is constant, as expected.
$h_0$
when the mobility is constant, as expected.

Figure 6.
 $(a)$
Scaling length
$(a)$
Scaling length
 $L_c$
as a function of time for tension-driven coarsening in a viscous film with varying
$L_c$
as a function of time for tension-driven coarsening in a viscous film with varying
 $h_0$
when
$h_0$
when
 $Q_{\gamma }=0.01$
.
$Q_{\gamma }=0.01$
.
 $L_c$
is calculated from the average
$L_c$
is calculated from the average
 $S(\boldsymbol {k},t)$
for
$S(\boldsymbol {k},t)$
for
 $N=22$
independent simulations. The inset shows the results when a constant mobility is used instead.
$N=22$
independent simulations. The inset shows the results when a constant mobility is used instead.
 $(b)$
Scaling length
$(b)$
Scaling length
 $L_c$
as a function of time for bending-driven coarsening in a viscous film with varying
$L_c$
as a function of time for bending-driven coarsening in a viscous film with varying
 $h_0$
when
$h_0$
when
 $Q_B=0.01$
.
$Q_B=0.01$
.
 $L_c$
is calculated from the average
$L_c$
is calculated from the average
 $S(\boldsymbol {k},t)$
for
$S(\boldsymbol {k},t)$
for
 $N=20$
independent simulations. The inset shows the results when a constant mobility is used instead.
$N=20$
independent simulations. The inset shows the results when a constant mobility is used instead.
In figure 6
 $(b)$
, similar results are presented, but this time, the tension term in (2.3) is replaced by the bending term, i.e. we set
$(b)$
, similar results are presented, but this time, the tension term in (2.3) is replaced by the bending term, i.e. we set
 $\gamma \gt 0$
and
$\gamma \gt 0$
and
 $B=0$
, and solve 2.8. As suggested by the theory in § 4.1, the power law for bending-driven coarsening is reduced from
$B=0$
, and solve 2.8. As suggested by the theory in § 4.1, the power law for bending-driven coarsening is reduced from
 $L_c\sim t^{1/3}$
. The inset in figure 6
$L_c\sim t^{1/3}$
. The inset in figure 6
 $(b)$
shows that this is indeed the case when the mobility is kept constant, with a growth exponent of approximately
$(b)$
shows that this is indeed the case when the mobility is kept constant, with a growth exponent of approximately
 $1/4$
consistently observed. For the viscous case, there is a decrease in the growth exponent when the value of
$1/4$
consistently observed. For the viscous case, there is a decrease in the growth exponent when the value of
 $h_0$
is increased, but this effect is not as pronounced as it is for the tension-driven films.
$h_0$
is increased, but this effect is not as pronounced as it is for the tension-driven films.
The decreased coarsening rate as
 $h_0$
is increased may seem counterintuitive at first glance, as a thicker film should lead to less viscous resistance to fluid flow and thus a higher mobility. In the coarsening process, however, the fluid flow between pockets must pass through the adhered region, where the film height is fixed at the equilibrium height of the adhesive molecules (
$h_0$
is increased may seem counterintuitive at first glance, as a thicker film should lead to less viscous resistance to fluid flow and thus a higher mobility. In the coarsening process, however, the fluid flow between pockets must pass through the adhered region, where the film height is fixed at the equilibrium height of the adhesive molecules (
 $h=1$
), thus restricting the mobility of the flow through this region. Increasing
$h=1$
), thus restricting the mobility of the flow through this region. Increasing
 $h_0$
thus only increases the depth of the pockets, which decreases the flux through the interface between the pocket and the attached region.
$h_0$
thus only increases the depth of the pockets, which decreases the flux through the interface between the pocket and the attached region.

Figure 7.
 $(a)$
Scaling length
$(a)$
Scaling length
 $L_c$
as a function of time for tension-driven coarsening in a viscous film with varying
$L_c$
as a function of time for tension-driven coarsening in a viscous film with varying
 $Q_{\gamma }$
. These results are for
$Q_{\gamma }$
. These results are for
 $h_0=1.25$
and the
$h_0=1.25$
and the
 $L_c$
is calculated from the average
$L_c$
is calculated from the average
 $S(\boldsymbol {k},t)$
for
$S(\boldsymbol {k},t)$
for
 $N=22$
independent simulations.
$N=22$
independent simulations.
 $(b)$
Scaling length
$(b)$
Scaling length
 $L_c$
as a function of time for bending-driven coarsening in a viscous film with varying
$L_c$
as a function of time for bending-driven coarsening in a viscous film with varying
 $Q_B$
. These results are for
$Q_B$
. These results are for
 $h_0=1.4$
and the
$h_0=1.4$
and the
 $L_c$
is calculated from the average
$L_c$
is calculated from the average
 $S(\boldsymbol {k},t)$
for
$S(\boldsymbol {k},t)$
for
 $N=15$
independent simulations.
$N=15$
independent simulations.
Figure 7 shows the coarsening behaviour for both tension-driven and bending-driven viscous films as
 $Q$
is varied by two orders of magnitude. In both cases, the value of
$Q$
is varied by two orders of magnitude. In both cases, the value of
 $L_c$
at the beginning of coarsening is larger when the fluctuations are increased to
$L_c$
at the beginning of coarsening is larger when the fluctuations are increased to
 $Q=0.5$
, due to increased coalescence at very early times. In the bending case, large fluctuations also lead to an earlier onset of the power-law coarsening regime, as can be seen in figure 7
$Q=0.5$
, due to increased coalescence at very early times. In the bending case, large fluctuations also lead to an earlier onset of the power-law coarsening regime, as can be seen in figure 7
 $(b)$
. During the late coarsening stage, however, fluctuations do not play a significant role. In figure 7
$(b)$
. During the late coarsening stage, however, fluctuations do not play a significant role. In figure 7
 $(a)$
, we observe that increasing the magnitude of fluctuations leads to a slight decrease in the power law for tension-dominated films, while figure 7
$(a)$
, we observe that increasing the magnitude of fluctuations leads to a slight decrease in the power law for tension-dominated films, while figure 7
 $(b)$
shows that changing
$(b)$
shows that changing
 $Q_B$
does not seem to significantly affect the power law of bending-dominated films. This seems to suggest that the main role of the fluctuations is simply to provide perturbations at early times which initiate the adhesion process.
$Q_B$
does not seem to significantly affect the power law of bending-dominated films. This seems to suggest that the main role of the fluctuations is simply to provide perturbations at early times which initiate the adhesion process.
5. Discussion
We have demonstrated that the waiting time for membranes to come into contact across a viscous channel can be effectively predicted by the rare-event theory. Although computational concerns limited the present study to 1-D films, the methods we present can be extended to two dimensions. In that case, the rare-event prediction will have the same form, but a modified prefactor. Our results demonstrate the utility of rare-event theory in predicting the average time for important but unlikely events to occur in stochastic systems. Such statistical techniques are relevant in many biological systems where rare events caused by stochastic fluctuations may be necessary for important biological processes (Chou & D’Orsogna Reference Chou and D’Orsogna2014). Although the adhesion of live cells often involves active cellular machinery such as actomyosin and filopodia to help cells bind, our results may be useful for estimating the likelihood that cells may come into contact when such active mechanisms are absent and may help to distinguish between actively and passively driven dynamics.
The membrane coarsening results of § 4 present a minimal mathematical description of coarsening behaviour similar to the recent experiments of Dinet et al. (Reference Dinet, Torres-Sánchez, Lanfranco, Di Michele, Arroyo and Staykova2023). In their experiments, an osmotic shock is applied to GUVs initially attached through biotin–neutravidin bonds to a supported lipid bilayer, pushing liquid from inside the GUV into the membrane–membrane channel, forming small unattached lumens. As the system evolves, the smaller lumens shrink and eventually disappear, while larger lumens grow. Our work suggests a physical model to explain the intrinsic coarsening process in those experiments. Based on the results of § 4, one would expect to observe power-law coarsening of the domains in such experiments. Unfortunately, the data in the experiments of Dinet et al. (Reference Dinet, Torres-Sánchez, Lanfranco, Di Michele, Arroyo and Staykova2023) are not quite amenable to study the coarsening law because the separate processes of fluid flux through the membrane into the channel, flux of fluid from pocket to pocket and flux of fluid from the pockets to the exterior region are all occurring simultaneously. Nevertheless, it points to experimental realisations allowing for the observation of the predicted power-law coarsening dynamics by isolating the interluminal coarsening process, e.g. by using a larger GUV so the centre of the adhesion patch is not as affected by flux to the outside.
Our work provides a framework for understanding coarsening exponents in thin films. The results of § 4.2 provide a scaling-based prediction that the power-law exponent is smaller for bending-driven than for tension-driven coarsening. A more rigorous prediction of the growth rate for bending-driven coarsening might be feasible applying renormalisation group theory, as is described by Bray (Reference Bray1994). Whether a physical system falls under the bending- or tension-dominated regime depends on the ratio of the characteristic length scale of the domains to the bendocapillary length scale,
 $L_{bc} = \sqrt {B/\gamma }$
. As such, there may be a cross-over between the two power-law regimes as the domain size grows past
$L_{bc} = \sqrt {B/\gamma }$
. As such, there may be a cross-over between the two power-law regimes as the domain size grows past
 $L_{bc}$
during coarsening. Observing this, however, would require simulations of growth across a larger range of length scales than that shown in this study. An estimate of
$L_{bc}$
during coarsening. Observing this, however, would require simulations of growth across a larger range of length scales than that shown in this study. An estimate of
 $L_{bc}$
based on typical values for biological membranes is smaller than the size of lumens in the experiments of Dinet et al. (Reference Dinet, Torres-Sánchez, Lanfranco, Di Michele, Arroyo and Staykova2023), which suggests that tension should dominate for that system. Even so, we would expect a growth exponent with time that is smaller than
$L_{bc}$
based on typical values for biological membranes is smaller than the size of lumens in the experiments of Dinet et al. (Reference Dinet, Torres-Sánchez, Lanfranco, Di Michele, Arroyo and Staykova2023), which suggests that tension should dominate for that system. Even so, we would expect a growth exponent with time that is smaller than
 ${\sim} t^{1/3}$
due to the nonlinear mobility coming from the fluid viscosity. It would also be interesting to see if the coarsening rate would be altered if one were to change the membrane properties to have a larger value of
${\sim} t^{1/3}$
due to the nonlinear mobility coming from the fluid viscosity. It would also be interesting to see if the coarsening rate would be altered if one were to change the membrane properties to have a larger value of
 $L_{bc}$
.
$L_{bc}$
.
The membrane deformation containing just the bending and isotropic tension terms (see § 2.2) is a simplification that ignores the fact that the tension will be affected by deformation (Landau & Lifshitz Reference Landau and Lifshitz1986; Peng & Lister Reference Peng and Lister2019). This allows us to characterise some aspects of how bending and isotropic tension act differently, but prevents us from getting a full understanding of how the deformation of a thin elastic sheet affects the dynamics of adhesion and coarsening. To complete the picture, the full Föppl–von Kármán equations could be implemented to properly describe stretching in the membrane (Lister, Peng & Neufeld Reference Lister, Peng and Neufeld2013; Pihler-Puzović et al. Reference Pihler-Puzović, Périllat, Russell, Juel and Heil2013).
Another observation to highlight is that the noise amplitude seems to have no effect on the power law for the coarsening process in both tension- and bending-dominated regimes. This is not true, however, for the spreading of droplets on a flat substrate, which can be seen as the coarsening of a single lumen. In the tension-dominated regime, as the droplet spreads, the growth of its radius follows the classical Tanner’s law (Tanner Reference Tanner1979) if the system is deterministic and follows a fluctuation-enhanced Tanner’s law (Davidovitch et al. Reference Davidovitch, Moro and Stone2005) in the presence of noise. In the bending-dominated regime, a different power law for growth was also found for deterministic (Lister et al. Reference Lister, Peng and Neufeld2013) and stochastic (Carlson Reference Carlson2018; Pedersen et al. Reference Pedersen, Niven, Salez, Dalnoki-Veress and Carlson2019) systems. One key difference between our set-up and droplet spreading is the presence of bound adhesive molecules, which keeps the membrane fixed at a constant height in the adhered region. Another difference is that instead of a single droplet, we have a network of pockets connected by adhesion patches.
Our numerical results predict that coarsening is slower for larger initial heights in viscous, tension-driven coarsening, which is directly relevant to coarsening in dewetting liquid films. Experiments on dewetting nanometric polymer films by Limary & Green (Reference Limary and Green2003) indeed showed that the coarsening exponent was smaller when the initial film height was increased. Although the driving potential in such a system comes from the surface energies of the materials rather than protein-like binding, the governing equations should otherwise be the same and we would expect similar dynamics. Further work on this problem could lead to a more complete understanding of how and why an increased initial film height delays coalescence.
Finally, a future interesting avenue to explore would be looking into experimental systems of elastohydrodynamic coarsening beyond a biological context. The predicted coarsening exponent (§ 4.1) is not specific to the molecular binding-based adhesive potential in our model, so similar coarsening dynamics should be observed even if the adhesive force in (2.4) is replaced by some other physical mechanism. For example, if liquid is trapped between two thin elastic sheets grafted with polymer brushes that are attracted to each other (Mani, Gopinath & Mahadevan Reference Mani, Gopinath and Mahadevan2012), one might observe coarsening of non-adhered pockets quite similar to the observations in our simulations.
Supplementary movies
Supplementary movies are available at https://doi.org/10.1017/jfm.2025.10872.
Acknowledgements
We thank Sami Al-Izzi for stimulating discussions and funding from the UiO:Life Science.
Declaration of interests
The authors report no conflict of interest.
Appendix A. Adhesive molecule kinetics
In this section, we present the simple model used for the kinetics of the adhesive molecule bond occuring across the intermembrane channel. In biological systems, this typically corresponds to the bond between two membrane-anchored proteins across the extracellular gap. The proteins are assumed to be present at all times on both sides of the channel at a constant concentration
 $c_0$
. The concentration of bound proteins at a particular location is denoted as
$c_0$
. The concentration of bound proteins at a particular location is denoted as
 $\hat {c}(\hat {x},\hat {y},\hat {t})$
. If proteins bind with a rate constant
$\hat {c}(\hat {x},\hat {y},\hat {t})$
. If proteins bind with a rate constant
 $K_{{on}}$
and unbind with a rate constant
$K_{{on}}$
and unbind with a rate constant
 $K_{\textit{off}}$
, the dynamics of
$K_{\textit{off}}$
, the dynamics of
 $\hat {c}(\hat {x},\hat {y},\hat {t})$
is governed by (assuming that diffusion and the flow are slow compared with the binding kinetics)
$\hat {c}(\hat {x},\hat {y},\hat {t})$
is governed by (assuming that diffusion and the flow are slow compared with the binding kinetics)
 \begin{equation} \frac {\partial \hat {c}}{\partial \hat {t}} = (c_0-\hat {c})K_{{on}}-\hat {c}K_{\textit{off}}. \end{equation}
\begin{equation} \frac {\partial \hat {c}}{\partial \hat {t}} = (c_0-\hat {c})K_{{on}}-\hat {c}K_{\textit{off}}. \end{equation}
The binding rate constant
 $K_{{on}}$
is represented as a probability of crossing an energy barrier and is dependent on the film height
$K_{{on}}$
is represented as a probability of crossing an energy barrier and is dependent on the film height
 $\hat {h}$
with a maximum likelihood of binding at
$\hat {h}$
with a maximum likelihood of binding at
 $\hat {h}=l$
(Bell et al. Reference Bell, Dembo and Bongrand1984). The off rate,
$\hat {h}=l$
(Bell et al. Reference Bell, Dembo and Bongrand1984). The off rate,
 $K_{\textit{off}}$
, is taken as a constant (Qi et al. Reference Qi, Groves and Chakraborty2001),
$K_{\textit{off}}$
, is taken as a constant (Qi et al. Reference Qi, Groves and Chakraborty2001),
 \begin{align} K_{{on}}=\frac {1}{\tau _{{on}}}\exp \left (-\left ((\hat {h}(\hat {x},\hat {y},\hat {t})-l)/\sigma \right)^2\right), && K_{\textit{off}}=\frac {1}{\tau _{\textit{off}}}\!, \end{align}
\begin{align} K_{{on}}=\frac {1}{\tau _{{on}}}\exp \left (-\left ((\hat {h}(\hat {x},\hat {y},\hat {t})-l)/\sigma \right)^2\right), && K_{\textit{off}}=\frac {1}{\tau _{\textit{off}}}\!, \end{align}
where
 $l$
is the equilibrium length of the intermembrane protein bond,
$l$
is the equilibrium length of the intermembrane protein bond,
 $\sigma$
is the width of the kinetic binding zone, and
$\sigma$
is the width of the kinetic binding zone, and
 $\tau _{{on}}$
and
$\tau _{{on}}$
and
 $\tau _{\textit{off}}$
are kinetic times.
$\tau _{\textit{off}}$
are kinetic times.
For this system, if the protein kinetics is allowed to relax much faster than the time scale of film deformation, we can assume the bound protein concentration
 $\hat {c}$
to be quasi-constant (Carlson & Mahadevan Reference Carlson and Mahadevan2015b
). At chemical equilibrium, (A1) leads us to express the equilibrium concentration
$\hat {c}$
to be quasi-constant (Carlson & Mahadevan Reference Carlson and Mahadevan2015b
). At chemical equilibrium, (A1) leads us to express the equilibrium concentration
 $\hat {c}^{eq}$
as
$\hat {c}^{eq}$
as
 \begin{equation} \hat {c}^{eq}(\hat {h}(\hat {x},\hat {y},\hat {t})) = c_0\frac { K_{{on}}}{ K_{{on}}+K_{\textit{off}}}, \end{equation}
\begin{equation} \hat {c}^{eq}(\hat {h}(\hat {x},\hat {y},\hat {t})) = c_0\frac { K_{{on}}}{ K_{{on}}+K_{\textit{off}}}, \end{equation}
which gives us the expression for
 $\hat {c}(\hat {h})$
in (2.5). In our simulations, we take the dimensionless binding distribution width
$\hat {c}(\hat {h})$
in (2.5). In our simulations, we take the dimensionless binding distribution width
 $\sigma /l$
to be 0.2, as was used by Carlson & Mahadevan (Reference Carlson and Mahadevan2015a
,Reference Carlson and Mahadevan
b
) to describe TCR–pMHC bonding in the immune synapse, although we note that defining precise values for this parameter is difficult and that higher values may be relevant (Qi et al. Reference Qi, Groves and Chakraborty2001). We use a value of
$\sigma /l$
to be 0.2, as was used by Carlson & Mahadevan (Reference Carlson and Mahadevan2015a
,Reference Carlson and Mahadevan
b
) to describe TCR–pMHC bonding in the immune synapse, although we note that defining precise values for this parameter is difficult and that higher values may be relevant (Qi et al. Reference Qi, Groves and Chakraborty2001). We use a value of
 $1/3$
for the binding/unbinding time scale ratio
$1/3$
for the binding/unbinding time scale ratio
 $\tau _{{on}}/\tau _{\textit{off}}$
. Although the values chosen for
$\tau _{{on}}/\tau _{\textit{off}}$
. Although the values chosen for
 $\sigma /l$
and
$\sigma /l$
and
 $\tau _{{on}}/\tau _{\textit{off}}$
are rather arbitrary, and not connected to any specific molecular interaction, we observe little change in our results when these parameters are altered. The same coarsening behaviour is observed as long as
$\tau _{{on}}/\tau _{\textit{off}}$
are rather arbitrary, and not connected to any specific molecular interaction, we observe little change in our results when these parameters are altered. The same coarsening behaviour is observed as long as
 $h_0$
is set just above
$h_0$
is set just above
 $1+\sigma /l$
.
$1+\sigma /l$
.
Finally, we note that the pressure contribution from the bound proteins, as described in (2.4) with
 $\hat {c}(\hat {h})$
given by (A3), can be written as the functional derivative of a free energy with respect to the film profile
$\hat {c}(\hat {h})$
given by (A3), can be written as the functional derivative of a free energy with respect to the film profile
 $\hat {h}(\hat {x},\hat {y})$
,
$\hat {h}(\hat {x},\hat {y})$
,
 \begin{equation} F_{\textit{adh}}[h] = \int - \frac { c_0\kappa \sigma ^2}{2}\left (\ln \left [\exp \left (-\frac {(\hat {h}(\hat {x},\hat {y},\hat {t})-l)^2}{\sigma ^2}\right)+\frac {\tau _{{on}}}{\tau _{\textit{off}}}\right ]\right)\,{\rm d}A. \end{equation}
\begin{equation} F_{\textit{adh}}[h] = \int - \frac { c_0\kappa \sigma ^2}{2}\left (\ln \left [\exp \left (-\frac {(\hat {h}(\hat {x},\hat {y},\hat {t})-l)^2}{\sigma ^2}\right)+\frac {\tau _{{on}}}{\tau _{\textit{off}}}\right ]\right)\,{\rm d}A. \end{equation}
This corresponds to a single energy well, as is shown in figure 8. Taking the functional derivative
 $\delta F_{adh}/\delta h$
gives the adhesion pressure term in (2.4) with the concentration given by (2.5).
$\delta F_{adh}/\delta h$
gives the adhesion pressure term in (2.4) with the concentration given by (2.5).

Figure 8. Plot of the energy density from (A4). The energy density
 $F$
is non-dimensionalised by
$F$
is non-dimensionalised by
 $c_0\kappa l^2$
and the height by
$c_0\kappa l^2$
and the height by
 $l$
. The binding distribution width is set to
$l$
. The binding distribution width is set to
 $\sigma /l=1/5$
and the time scale ratio is
$\sigma /l=1/5$
and the time scale ratio is
 $\tau _{{on}}/\tau _{\textit{off}}=1/3$
.
$\tau _{{on}}/\tau _{\textit{off}}=1/3$
.
Appendix B. Non-dimensionalisation of equations
As mentioned in § 2.5, we non-dimensionalise the governing equations before computing our numerical solutions. In this section, we provide the details of how we have done so for each of the cases discussed in §§ 3 and 4.
B.1. Attachment simulations
For the simulations to estimate the binding time of a fluctuating film, a quasi-1-D domain of length
 $L$
in the x-direction and width
$L$
in the x-direction and width
 $w$
in the y-direction is considered. Since we are focused on the effects of bending, tension is not included. We also ignore the protein-binding term since we are interested in how long it takes to reach the values of
$w$
in the y-direction is considered. Since we are focused on the effects of bending, tension is not included. We also ignore the protein-binding term since we are interested in how long it takes to reach the values of
 $h$
where these become significant (similar results are observed when they are included, but the theoretical calculation is more challenging). Under these circumstances and considering the bending-dominated case, the 1-D version of (2.1) simplifies to
$h$
where these become significant (similar results are observed when they are included, but the theoretical calculation is more challenging). Under these circumstances and considering the bending-dominated case, the 1-D version of (2.1) simplifies to
 \begin{equation} \frac {\partial \hat {h}(\hat {x},\hat {t})}{\partial \hat {t}} = \frac {\partial }{\partial \hat {x}} \left (\frac {\hat {h}(\hat {\hat {x}},\hat {t})^3}{12\mu }\frac {\partial }{\partial \hat {x}} \left (B\frac {\partial ^4}{\partial \hat {x}^4} \hat {h}(\hat {x},\hat {t})\right)\right) + \sqrt {\frac {k_B T}{6\mu w }}\frac {\partial }{\partial \hat {x}} \big (\hat {h}(\hat {x},\hat {t})^{3/2}\hat {\eta }(\hat {x},\hat {t})\big). \end{equation}
\begin{equation} \frac {\partial \hat {h}(\hat {x},\hat {t})}{\partial \hat {t}} = \frac {\partial }{\partial \hat {x}} \left (\frac {\hat {h}(\hat {\hat {x}},\hat {t})^3}{12\mu }\frac {\partial }{\partial \hat {x}} \left (B\frac {\partial ^4}{\partial \hat {x}^4} \hat {h}(\hat {x},\hat {t})\right)\right) + \sqrt {\frac {k_B T}{6\mu w }}\frac {\partial }{\partial \hat {x}} \big (\hat {h}(\hat {x},\hat {t})^{3/2}\hat {\eta }(\hat {x},\hat {t})\big). \end{equation}
The finite width
 $w$
must be included here since thermal flucutations are inherently a 3-D phenomenon, but will in the end be absorbed into the prefactor of the stochastic term (Grün et al. Reference Grün, Mecke and Rauscher2006). We non-dimensionalise (B1) by introducing the scaling relations:
$w$
must be included here since thermal flucutations are inherently a 3-D phenomenon, but will in the end be absorbed into the prefactor of the stochastic term (Grün et al. Reference Grün, Mecke and Rauscher2006). We non-dimensionalise (B1) by introducing the scaling relations:
 \begin{align} \hat {h}=hh_0, \hspace {10pt} \hat {x}=xL \hspace {10pt} \hat {t}=t\frac {12 \mu L^6}{Bh_0^3}, \hspace {10pt}\hat {\eta }=\eta \sqrt {\frac {B h_0^3}{12 \mu L^7}}, \end{align}
\begin{align} \hat {h}=hh_0, \hspace {10pt} \hat {x}=xL \hspace {10pt} \hat {t}=t\frac {12 \mu L^6}{Bh_0^3}, \hspace {10pt}\hat {\eta }=\eta \sqrt {\frac {B h_0^3}{12 \mu L^7}}, \end{align}
where the dimensionless variables are without hats. The time is scaled by a characteristic time scale on which bent elastohydrodynamic thin films relax (Carlson Reference Carlson2018) (which can be obtained by scaling the left-hand side of (B1) with the first term on the right-hand side), and
 $\boldsymbol{\eta }$
is scaled by a dimensionally correct combination of the time and horizontal length scales. When these relations are inserted into (B1), the dimensionless thin-film equation becomes
$\boldsymbol{\eta }$
is scaled by a dimensionally correct combination of the time and horizontal length scales. When these relations are inserted into (B1), the dimensionless thin-film equation becomes
 \begin{equation} \frac {\partial h}{\partial t} = \frac {\partial }{\partial x} \left (h^3\frac {\partial }{\partial x} \left (\frac {\partial ^4}{\partial x^4} h\right)\right) + Q_{\text{1-D}}\frac {\partial }{\partial x} \big (h^{3/2}\eta \big). \end{equation}
\begin{equation} \frac {\partial h}{\partial t} = \frac {\partial }{\partial x} \left (h^3\frac {\partial }{\partial x} \left (\frac {\partial ^4}{\partial x^4} h\right)\right) + Q_{\text{1-D}}\frac {\partial }{\partial x} \big (h^{3/2}\eta \big). \end{equation}
The non-dimensional number
 $Q_{\text{1-D}}= ({L}/{h_0})\sqrt { {2k_B T L}/{Bw}}$
represents the strength of the thermal fluctuations in the domain. In fact,
$Q_{\text{1-D}}= ({L}/{h_0})\sqrt { {2k_B T L}/{Bw}}$
represents the strength of the thermal fluctuations in the domain. In fact,
 $Q_{\text{1-D}}$
is directly proportional to the thermal roughness of the film by the relation
$Q_{\text{1-D}}$
is directly proportional to the thermal roughness of the film by the relation
 $Q_{\text{1-D}}=(\langle |\delta \hat {h}|\rangle /h_0)(360)^{-1/2}$
, where
$Q_{\text{1-D}}=(\langle |\delta \hat {h}|\rangle /h_0)(360)^{-1/2}$
, where
 $\langle |\delta \hat {h}|\rangle$
is the thermal roughness of a film.
$\langle |\delta \hat {h}|\rangle$
is the thermal roughness of a film.
B.2. Coarsening simulations
For the simulations of coarsening in thin films, a 2-D domain was used and the non-dimensionalisation of the equations is different as opposed to the attachment simulations. In this section, we decribe how (2.1) is non-dimensionalised when the protein dynamics of (2.4) is included. The approach is different in the bending-driven and tension-driven cases because in each case, the horizontal length scale is non-dimensionalised by the characteristic domain size obtained by balancing the protein forces with the driving membrane force (Carlson & Mahadevan Reference Carlson and Mahadevan2015a ).
B.2.1. Bending driven
When coarsening is driven by bending, (2.1) becomes
 \begin{equation} \frac {\partial \hat {h}}{\partial \hat {t}} = \hat {\boldsymbol{\nabla }}\boldsymbol{\cdot }\left (\frac {\hat {h}^3}{12\mu }\hat {\boldsymbol{\nabla }} \left (B\hat {{\nabla} }^4\hat {h}+\kappa (\hat {h}-l)c\right) \right) + \sqrt {\frac {k_B T}{6\mu }}\hat {\boldsymbol{\nabla }} \boldsymbol{\cdot }\big (\hat {h}^{3/2}\boldsymbol{\hat {\eta }}\big). \end{equation}
\begin{equation} \frac {\partial \hat {h}}{\partial \hat {t}} = \hat {\boldsymbol{\nabla }}\boldsymbol{\cdot }\left (\frac {\hat {h}^3}{12\mu }\hat {\boldsymbol{\nabla }} \left (B\hat {{\nabla} }^4\hat {h}+\kappa (\hat {h}-l)c\right) \right) + \sqrt {\frac {k_B T}{6\mu }}\hat {\boldsymbol{\nabla }} \boldsymbol{\cdot }\big (\hat {h}^{3/2}\boldsymbol{\hat {\eta }}\big). \end{equation}
We non-dimensionalise (B4) by introducing the scaling relations
 \begin{align} &\hat {h}=hl, \quad \hat {x}=x\left (\frac {B}{c_0\kappa }\right)^{1/4}, \quad \hat {y}=y\left (\frac {B}{c_0\kappa }\right)^{1/4}, \notag\\ &\hat {t}=t\frac {12\mu B^{1/2}}{l^3(c_0\kappa)^{3/2}}, \quad \hat {c}=cc_0, \quad \boldsymbol{\hat {\eta }}=\boldsymbol{\eta }\frac {c_0\kappa l^{3/2}}{(12B\mu)^{1/2}} ,\end{align}
\begin{align} &\hat {h}=hl, \quad \hat {x}=x\left (\frac {B}{c_0\kappa }\right)^{1/4}, \quad \hat {y}=y\left (\frac {B}{c_0\kappa }\right)^{1/4}, \notag\\ &\hat {t}=t\frac {12\mu B^{1/2}}{l^3(c_0\kappa)^{3/2}}, \quad \hat {c}=cc_0, \quad \boldsymbol{\hat {\eta }}=\boldsymbol{\eta }\frac {c_0\kappa l^{3/2}}{(12B\mu)^{1/2}} ,\end{align}
where the dimensionless variables are without hats. The characteristic horizontal length scale by which
 $x$
and
$x$
and
 $y$
are non-dimensionalised,
$y$
are non-dimensionalised,
 $(B/c_0\kappa)^{1/4}$
, comes from a balance between the protein spring pressure and the bending pressure, and describes the characteristic length scale of protein domains (Carlson & Mahadevan Reference Carlson and Mahadevan2015a
). The time scale comes from a balance of the left-hand side term in (B4) with the bending pressure term, using the aforementioned horizontal length scale to scale the gradient
$(B/c_0\kappa)^{1/4}$
, comes from a balance between the protein spring pressure and the bending pressure, and describes the characteristic length scale of protein domains (Carlson & Mahadevan Reference Carlson and Mahadevan2015a
). The time scale comes from a balance of the left-hand side term in (B4) with the bending pressure term, using the aforementioned horizontal length scale to scale the gradient
 $\hat {\boldsymbol{\nabla }}$
. When these scalings are introduced into (B6), the dimensionless thin film equation becomes
$\hat {\boldsymbol{\nabla }}$
. When these scalings are introduced into (B6), the dimensionless thin film equation becomes
 \begin{equation} \frac {\partial h}{\partial t} = \boldsymbol{\nabla }\boldsymbol{\cdot }\big (h^3\boldsymbol{\nabla }\big ({\nabla} ^4h+ (h-1)c\big) \big) + Q_{B}\boldsymbol{\nabla }\boldsymbol{\cdot }\big (h^{3/2}\boldsymbol{\eta }\big). \end{equation}
\begin{equation} \frac {\partial h}{\partial t} = \boldsymbol{\nabla }\boldsymbol{\cdot }\big (h^3\boldsymbol{\nabla }\big ({\nabla} ^4h+ (h-1)c\big) \big) + Q_{B}\boldsymbol{\nabla }\boldsymbol{\cdot }\big (h^{3/2}\boldsymbol{\eta }\big). \end{equation}
The non-dimensional number
 $Q_{B}=({1}/{l})\sqrt {{2k_B T }/{B^{1/2}(\kappa c_0)^{1/2}}}$
represents the strength of the thermal fluctuations in the domain. In fact,
$Q_{B}=({1}/{l})\sqrt {{2k_B T }/{B^{1/2}(\kappa c_0)^{1/2}}}$
represents the strength of the thermal fluctuations in the domain. In fact,
 $Q_{B}$
is directly proportional to the average amplitude of thermal fluctuations of the film by the relation
$Q_{B}$
is directly proportional to the average amplitude of thermal fluctuations of the film by the relation
 $Q_{B}\sim (\langle |\delta \hat {h}|\rangle /l)((B/(c_0\kappa))^{1/4}/L)$
, where
$Q_{B}\sim (\langle |\delta \hat {h}|\rangle /l)((B/(c_0\kappa))^{1/4}/L)$
, where
 $\langle |\delta \hat {h}|\rangle$
is the thermal roughness of a film. A physical interpretation for
$\langle |\delta \hat {h}|\rangle$
is the thermal roughness of a film. A physical interpretation for
 $Q_{B}$
is thus a non-dimensionless thermal roughness of the film, although the amplitude is dependent on the domain size, which is an inherent and poorly studied feature of thermal fluctuations in such films.
$Q_{B}$
is thus a non-dimensionless thermal roughness of the film, although the amplitude is dependent on the domain size, which is an inherent and poorly studied feature of thermal fluctuations in such films.
B.2.2. Tension-driven
When coarsening is driven by tension, (2.1) becomes
 \begin{equation} \frac {\partial \hat {h}}{\partial \hat {t}} = \hat {\boldsymbol{\nabla }}\boldsymbol{\cdot }\left (\frac {\hat {h}^3}{12\mu }\hat {\boldsymbol{\nabla }} \big (-\gamma \hat {{\nabla} }^2\hat {h}+\kappa (\hat {h}-l)\hat {c}\big) \right) + \sqrt {\frac {k_B T}{6\mu }}\hat {\boldsymbol{\nabla }} \boldsymbol{\cdot }\big (\hat {h}^{3/2}\boldsymbol{\hat {\eta }}\big) . \end{equation}
\begin{equation} \frac {\partial \hat {h}}{\partial \hat {t}} = \hat {\boldsymbol{\nabla }}\boldsymbol{\cdot }\left (\frac {\hat {h}^3}{12\mu }\hat {\boldsymbol{\nabla }} \big (-\gamma \hat {{\nabla} }^2\hat {h}+\kappa (\hat {h}-l)\hat {c}\big) \right) + \sqrt {\frac {k_B T}{6\mu }}\hat {\boldsymbol{\nabla }} \boldsymbol{\cdot }\big (\hat {h}^{3/2}\boldsymbol{\hat {\eta }}\big) . \end{equation}
We non-dimensionalise (B7) by introducing the scaling relations
 \begin{align} &\hat {h}=hl, \quad \hat {x}=x\left (\frac {\gamma }{c_0\kappa }\right)^{1/2}, \quad \hat {y}=y\left (\frac {\gamma }{c_0\kappa }\right)^{1/2}, \notag\\ &\hat {t}=t\frac {12\mu \gamma }{l^3(c_0\kappa)^{2}}, \quad \hat {c}=cc_0, \quad \boldsymbol{\hat {\eta }}=\boldsymbol{\eta }\frac {(c_0\kappa)^{3/2} l^{3/2}}{(12\mu)^{1/2}\gamma }, \end{align}
\begin{align} &\hat {h}=hl, \quad \hat {x}=x\left (\frac {\gamma }{c_0\kappa }\right)^{1/2}, \quad \hat {y}=y\left (\frac {\gamma }{c_0\kappa }\right)^{1/2}, \notag\\ &\hat {t}=t\frac {12\mu \gamma }{l^3(c_0\kappa)^{2}}, \quad \hat {c}=cc_0, \quad \boldsymbol{\hat {\eta }}=\boldsymbol{\eta }\frac {(c_0\kappa)^{3/2} l^{3/2}}{(12\mu)^{1/2}\gamma }, \end{align}
where the dimensionless variables are without hats. The characteristic horizontal length scale by which
 $x$
and
$x$
and
 $y$
are non-dimensionalised,
$y$
are non-dimensionalised,
 $(\gamma /c_0\kappa)^{1/2}$
, comes from a balance between the protein spring pressure and the interfacial tension, and describes the characteristic length scale of protein domains. The time scale comes from a balance of the left-hand side term in (B7) with the tension term, using the aforementioned horizontal length scale to scale the gradient
$(\gamma /c_0\kappa)^{1/2}$
, comes from a balance between the protein spring pressure and the interfacial tension, and describes the characteristic length scale of protein domains. The time scale comes from a balance of the left-hand side term in (B7) with the tension term, using the aforementioned horizontal length scale to scale the gradient
 $\hat {\boldsymbol{\nabla }}$
. When these scalings are introduced into (B7), the dimensionless thin film equation becomes
$\hat {\boldsymbol{\nabla }}$
. When these scalings are introduced into (B7), the dimensionless thin film equation becomes
 \begin{equation} \frac {\partial h}{\partial t} = \boldsymbol{\nabla }\boldsymbol{\cdot }\big (h^3\boldsymbol{\nabla }\big (-{\nabla} ^2h+ (h-1)c\big) \big) + Q_{\gamma }\boldsymbol{\nabla }\boldsymbol{\cdot }\big (h^{3/2}\boldsymbol{\eta }\big). \end{equation}
\begin{equation} \frac {\partial h}{\partial t} = \boldsymbol{\nabla }\boldsymbol{\cdot }\big (h^3\boldsymbol{\nabla }\big (-{\nabla} ^2h+ (h-1)c\big) \big) + Q_{\gamma }\boldsymbol{\nabla }\boldsymbol{\cdot }\big (h^{3/2}\boldsymbol{\eta }\big). \end{equation}
The non-dimensional number
 $Q_{\gamma }= ({1}/{l})\sqrt { {2k_B T }/{\gamma }}$
represents the strength of the thermal fluctuations in the domain. In fact,
$Q_{\gamma }= ({1}/{l})\sqrt { {2k_B T }/{\gamma }}$
represents the strength of the thermal fluctuations in the domain. In fact,
 $Q_{\gamma }$
is directly proportional to the average amplitude of thermal fluctuations of a freely fluctuating film subjected to interfacial tension by the relation
$Q_{\gamma }$
is directly proportional to the average amplitude of thermal fluctuations of a freely fluctuating film subjected to interfacial tension by the relation
 $Q_{\gamma }\sim (\langle |\delta \hat {h}|\rangle /l)$
, where
$Q_{\gamma }\sim (\langle |\delta \hat {h}|\rangle /l)$
, where
 $\langle |\delta \hat {h}|\rangle$
is the thermal roughness of a film without the protein binding term. A physical interpretation for
$\langle |\delta \hat {h}|\rangle$
is the thermal roughness of a film without the protein binding term. A physical interpretation for
 $Q_{\gamma }$
is thus a non-dimensionless thermal roughness of the film, which for a tension-dominated film is not dependent on
$Q_{\gamma }$
is thus a non-dimensionless thermal roughness of the film, which for a tension-dominated film is not dependent on
 $L$
. This has in fact been verified numerically in a previous work where the length scale
$L$
. This has in fact been verified numerically in a previous work where the length scale
 $\langle |\delta \hat {h}|\rangle$
was calculated directly (Dhaliwal et al. Reference Dhaliwal, Pedersen, Kadri, Miquelard-Garnier, Sollogoub, Peixinho, Salez and Carlson2024).
$\langle |\delta \hat {h}|\rangle$
was calculated directly (Dhaliwal et al. Reference Dhaliwal, Pedersen, Kadri, Miquelard-Garnier, Sollogoub, Peixinho, Salez and Carlson2024).
Appendix C. Rare-event theory
To predict the average waiting time (mean first passage time) for proteins to bind, we apply the rare-event theory for a gradient flow following the procedure outlined by Liu et al. (Reference Liu, Sprittles and Grafke2024), with a simple modification in the asymptotic since the system is not bistable, that is, the transition is not from one local minimum to another. For the mean first passage time of a non-gradient system, the reader can refer to Grafke, Schäfer & Vanden-Eijnden (Reference Grafke, Schäfer and Vanden-Eijnden2024). To make the derivation more general and easier to follow, we will first derive the formula of the mean first passage time for a general gradient flow and then demonstrate the application to the elastohydrodynamic thin film equation.
C.1. Mean first passage time for gradient flow
Let us first formalise the problem. Consider a general stochastic differential equation describing a gradient flow
 \begin{equation} {\rm d}X_t = -M(X_t)\boldsymbol{\nabla }F(X_t) \,{\rm d}t + \sqrt {2\varepsilon } M_{1/2}(X_t)\,{\rm d}W_t\,, \end{equation}
\begin{equation} {\rm d}X_t = -M(X_t)\boldsymbol{\nabla }F(X_t) \,{\rm d}t + \sqrt {2\varepsilon } M_{1/2}(X_t)\,{\rm d}W_t\,, \end{equation}
where
 $X_t\in \mathbb{R}^{n}$
is a random variable,
$X_t\in \mathbb{R}^{n}$
is a random variable,
 $M(X_t):\mathbb{R}^n\to \mathbb{R}^{n\times n}$
is the semi-positive definite mobility matrix (or mobility operator),
$M(X_t):\mathbb{R}^n\to \mathbb{R}^{n\times n}$
is the semi-positive definite mobility matrix (or mobility operator),
 $F(X_t)$
is some free energy of the system,
$F(X_t)$
is some free energy of the system,
 $\boldsymbol{\nabla}$
is the gradient with respect to
$\boldsymbol{\nabla}$
is the gradient with respect to
 $X_t$
,
$X_t$
,
 $\varepsilon$
is the noise amplitude,
$\varepsilon$
is the noise amplitude,
 $M_{1/2}(X_t):\mathbb{R}^{n}\to \mathbb{R}^{n\times n}$
is the ‘square root’ of the mobility, namely
$M_{1/2}(X_t):\mathbb{R}^{n}\to \mathbb{R}^{n\times n}$
is the ‘square root’ of the mobility, namely
 $M_{1/2}M^{T}_{1/2}=M$
(
$M_{1/2}M^{T}_{1/2}=M$
(
 $^{T}$
stands for transpose or Hermitian adjoint), and
$^{T}$
stands for transpose or Hermitian adjoint), and
 $W_t$
is the
$W_t$
is the
 $n$
-dimensional Brownian motion. It describes a system driven by the negative gradient of energy, i.e. a force that minimises the energy locally, while being disturbed by a Gaussian white noise. For simplicity, let
$n$
-dimensional Brownian motion. It describes a system driven by the negative gradient of energy, i.e. a force that minimises the energy locally, while being disturbed by a Gaussian white noise. For simplicity, let
 $-M(X_t)\boldsymbol{\nabla }F(X_t)= b(X_t)$
and
$-M(X_t)\boldsymbol{\nabla }F(X_t)= b(X_t)$
and
 $\sqrt {2}M_{1/2}(X_t)=\xi (X_t)$
, then (C1) is
$\sqrt {2}M_{1/2}(X_t)=\xi (X_t)$
, then (C1) is
 \begin{equation} {\rm d}X_t = b(X_t)\,{\rm d}t + \sqrt {\varepsilon }\xi (X_t)\,{\rm d}W_t\,. \end{equation}
\begin{equation} {\rm d}X_t = b(X_t)\,{\rm d}t + \sqrt {\varepsilon }\xi (X_t)\,{\rm d}W_t\,. \end{equation}
Assume there is a stable fixed point
 $x_A$
such that
$x_A$
such that
 $b(x_A)=0$
, and the time it takes for the system to first exit a basin of attraction
$b(x_A)=0$
, and the time it takes for the system to first exit a basin of attraction
 $D$
of
$D$
of
 $x_A$
starting at
$x_A$
starting at
 $x\in D$
is denoted as
$x\in D$
is denoted as
 \begin{equation} T_B(x) = \inf \{t\gt 0|X_t\notin D\}. \end{equation}
\begin{equation} T_B(x) = \inf \{t\gt 0|X_t\notin D\}. \end{equation}
We are interested in the the mean first passage time,
 $w_B(x)=\mathbb{E}[T_B(x)]$
, that is, the expectation of
$w_B(x)=\mathbb{E}[T_B(x)]$
, that is, the expectation of
 $T_B$
, which fulfils the inhomogeneous stationary Kolmogorov (Gardiner Reference Gardiner2009)
$T_B$
, which fulfils the inhomogeneous stationary Kolmogorov (Gardiner Reference Gardiner2009)
 \begin{equation} \begin{cases} \mathcal{L}w_B(x)=-1& \text{for } x\in D,\\ w_B(x)=0 & \text{for } x\in \partial D, \end{cases} \end{equation}
\begin{equation} \begin{cases} \mathcal{L}w_B(x)=-1& \text{for } x\in D,\\ w_B(x)=0 & \text{for } x\in \partial D, \end{cases} \end{equation}
where
 $\partial D$
is the boundary of the basin of attraction
$\partial D$
is the boundary of the basin of attraction
 $D$
. Here, we choose
$D$
. Here, we choose
 $D$
such that there is a global minimum
$D$
such that there is a global minimum
 $x_B$
of
$x_B$
of
 $F(x)$
on
$F(x)$
on
 $\partial D$
, and the normal vector
$\partial D$
, and the normal vector
 $\hat n$
of
$\hat n$
of
 $\partial D$
pointing outwards at
$\partial D$
pointing outwards at
 $x_B$
aligns with
$x_B$
aligns with
 $\boldsymbol{\nabla }F$
, that is,
$\boldsymbol{\nabla }F$
, that is,
 $\partial D$
is tangent to the contour line of the energy landscape only at
$\partial D$
is tangent to the contour line of the energy landscape only at
 $x_B$
. Here,
$x_B$
. Here,
 $\mathcal{L}$
is the generator of (C2),
$\mathcal{L}$
is the generator of (C2),
 \begin{equation} \mathcal{L} = b(x)\boldsymbol{\cdot }\boldsymbol{\nabla }+\frac {1}{2}\varepsilon a(x):\boldsymbol{\nabla }\boldsymbol{\nabla }, \end{equation}
\begin{equation} \mathcal{L} = b(x)\boldsymbol{\cdot }\boldsymbol{\nabla }+\frac {1}{2}\varepsilon a(x):\boldsymbol{\nabla }\boldsymbol{\nabla }, \end{equation}
where
 $a(x) = \varepsilon \xi (x)\xi ^{T}(x)$
is the noise covariance matrix and: is the scalar product. From the generator, we can deduce the invariant distribution
$a(x) = \varepsilon \xi (x)\xi ^{T}(x)$
is the noise covariance matrix and: is the scalar product. From the generator, we can deduce the invariant distribution
 $\rho _\infty (x)$
through the stationary Fokker–Planck equation
$\rho _\infty (x)$
through the stationary Fokker–Planck equation
 \begin{equation} \mathcal{L}^\dagger \rho _\infty =0, \end{equation}
\begin{equation} \mathcal{L}^\dagger \rho _\infty =0, \end{equation}
where
 $\mathcal{L}^\dagger$
is the
$\mathcal{L}^\dagger$
is the
 $L^2$
-adjoint of the generator
$L^2$
-adjoint of the generator
 \begin{equation} \mathcal{L}^\dagger \circ =-\boldsymbol{\nabla }\boldsymbol{\cdot }(b(x)\,\circ)+\frac {1}{2}\varepsilon \boldsymbol{\nabla }\boldsymbol{\nabla }:(a(x)\,\circ). \end{equation}
\begin{equation} \mathcal{L}^\dagger \circ =-\boldsymbol{\nabla }\boldsymbol{\cdot }(b(x)\,\circ)+\frac {1}{2}\varepsilon \boldsymbol{\nabla }\boldsymbol{\nabla }:(a(x)\,\circ). \end{equation}
In the case of a gradient flow (C1), one can show that the invariant distribution is given as the Gibbs distribution
 \begin{equation} \rho _\infty (x) = C e^{-F(x)/\varepsilon }, \end{equation}
\begin{equation} \rho _\infty (x) = C e^{-F(x)/\varepsilon }, \end{equation}
where
 $C$
is some constant. This will be used later in calculating the mean first passage time, which becomes significantly more complex if a form other than the Gibbs distribution is used. From the large deviation theory (Freidlin, Szucs & Wentzell Reference Freidlin, Szucs and Wentzell2012), we know that for
$C$
is some constant. This will be used later in calculating the mean first passage time, which becomes significantly more complex if a form other than the Gibbs distribution is used. From the large deviation theory (Freidlin, Szucs & Wentzell Reference Freidlin, Szucs and Wentzell2012), we know that for
 $\varepsilon \to 0$
,
$\varepsilon \to 0$
,
 \begin{equation} w_B(x_A)\asymp e^{\Delta F/\varepsilon }, \end{equation}
\begin{equation} w_B(x_A)\asymp e^{\Delta F/\varepsilon }, \end{equation}
where
 $\Delta F=(F(x_B)-F(x_A))$
. In other words, for the limiting case of small noise amplitude
$\Delta F=(F(x_B)-F(x_A))$
. In other words, for the limiting case of small noise amplitude
 $\varepsilon$
, the process almost certainly exits the basin of attraction
$\varepsilon$
, the process almost certainly exits the basin of attraction
 $D$
at the saddle point
$D$
at the saddle point
 $x_B$
, and the mean first passage time scales exponentially with the energy barrier between
$x_B$
, and the mean first passage time scales exponentially with the energy barrier between
 $x_A$
and
$x_A$
and
 $x_B$
with some unknown prefactor. To calculate the prefactor, we define a new random variable
$x_B$
with some unknown prefactor. To calculate the prefactor, we define a new random variable
 \begin{equation} \tau (x) = e^{-\Delta F/\varepsilon } w_B(x), \end{equation}
\begin{equation} \tau (x) = e^{-\Delta F/\varepsilon } w_B(x), \end{equation}
and the Kolmogorov (C4) becomes
 \begin{equation} \begin{cases} \mathcal{L}\tau (x)=-e^{-\Delta F/\varepsilon }& \text{for } x\in D,\\ \tau (x)=0 & \text{for } x\in \partial D. \end{cases} \end{equation}
\begin{equation} \begin{cases} \mathcal{L}\tau (x)=-e^{-\Delta F/\varepsilon }& \text{for } x\in D,\\ \tau (x)=0 & \text{for } x\in \partial D. \end{cases} \end{equation}
Consider a point
 $x\in D$
near the boundary
$x\in D$
near the boundary
 $\partial D$
, we can expand asymptotically in the direction of
$\partial D$
, we can expand asymptotically in the direction of
 $\hat n$
$\hat n$
 \begin{equation} x = u - \varepsilon \eta \hat n, \end{equation}
\begin{equation} x = u - \varepsilon \eta \hat n, \end{equation}
where
 $u\in \partial D$
and
$u\in \partial D$
and
 $\eta \gt 0$
. Note that this is different from the asymptotic expansion used by Liu et al. (Reference Liu, Sprittles and Grafke2024). This expansion leads to
$\eta \gt 0$
. Note that this is different from the asymptotic expansion used by Liu et al. (Reference Liu, Sprittles and Grafke2024). This expansion leads to
 \begin{align} \boldsymbol{\nabla }f &= \frac {\partial f}{\partial x_i} \boldsymbol{e}_i = \frac {{\rm d} f}{{\rm d} \eta }\frac {\partial \eta }{\partial x_i}\boldsymbol{e}_i = -\frac {1}{\varepsilon }\frac {{\rm d}f}{{\rm d}\eta }\hat n, \end{align}
\begin{align} \boldsymbol{\nabla }f &= \frac {\partial f}{\partial x_i} \boldsymbol{e}_i = \frac {{\rm d} f}{{\rm d} \eta }\frac {\partial \eta }{\partial x_i}\boldsymbol{e}_i = -\frac {1}{\varepsilon }\frac {{\rm d}f}{{\rm d}\eta }\hat n, \end{align}
 \begin{align} a:\boldsymbol{\nabla }\boldsymbol{\nabla }f &= a_{\textit{ij}}\frac {\partial }{\partial x_i}\frac {\partial }{\partial x_j}f = \frac {1}{\varepsilon ^2}a_{ij}\frac {\textrm{d}^2 f}{{\rm d}\eta ^2}\hat n_i\hat n_j = \frac {1}{\varepsilon ^2}\hat n\boldsymbol{\cdot }(a\hat n)\frac {\textrm{d}^2 f}{{\rm d}\eta ^2}. \end{align}
\begin{align} a:\boldsymbol{\nabla }\boldsymbol{\nabla }f &= a_{\textit{ij}}\frac {\partial }{\partial x_i}\frac {\partial }{\partial x_j}f = \frac {1}{\varepsilon ^2}a_{ij}\frac {\textrm{d}^2 f}{{\rm d}\eta ^2}\hat n_i\hat n_j = \frac {1}{\varepsilon ^2}\hat n\boldsymbol{\cdot }(a\hat n)\frac {\textrm{d}^2 f}{{\rm d}\eta ^2}. \end{align}
We can then expand the generator to the leading order and rewrite (C5) as
 \begin{align} \notag \mathcal{L} &= b(x)\boldsymbol{\cdot }\boldsymbol{\nabla }+ \frac {1}{2}\varepsilon a(x):\boldsymbol{\nabla }\boldsymbol{\nabla }\approx b(u)\boldsymbol{\cdot }\boldsymbol{\nabla }+ \frac {1}{2}\varepsilon a(u) :\boldsymbol{\nabla }\boldsymbol{\nabla }\\ &=\frac {1}{\varepsilon }\Big [ \underbrace {-b(u)\boldsymbol{\cdot }\hat n(u)}_{\beta (u)}\frac {\rm d}{{\rm d}\eta }+\underbrace {\frac {1}{2}\hat n \boldsymbol{\cdot }(a(u)\hat n)}_{\alpha (u)}\frac {\textrm{d}^2}{{\rm d}\eta ^2} \Big ]. \end{align}
\begin{align} \notag \mathcal{L} &= b(x)\boldsymbol{\cdot }\boldsymbol{\nabla }+ \frac {1}{2}\varepsilon a(x):\boldsymbol{\nabla }\boldsymbol{\nabla }\approx b(u)\boldsymbol{\cdot }\boldsymbol{\nabla }+ \frac {1}{2}\varepsilon a(u) :\boldsymbol{\nabla }\boldsymbol{\nabla }\\ &=\frac {1}{\varepsilon }\Big [ \underbrace {-b(u)\boldsymbol{\cdot }\hat n(u)}_{\beta (u)}\frac {\rm d}{{\rm d}\eta }+\underbrace {\frac {1}{2}\hat n \boldsymbol{\cdot }(a(u)\hat n)}_{\alpha (u)}\frac {\textrm{d}^2}{{\rm d}\eta ^2} \Big ]. \end{align}
As
 $\varepsilon \to 0$
, the leading term of the right-hand side of the Kolmogorov (C11) is zero, and we arrive at
$\varepsilon \to 0$
, the leading term of the right-hand side of the Kolmogorov (C11) is zero, and we arrive at
 \begin{equation} 0 = \mathcal{L}\tau (\eta) = \frac {1}{\varepsilon }\Big [\beta (u)\frac {\partial \tau }{\partial \eta }+\alpha (u)\frac {\partial ^2\tau }{\partial \eta ^2}\Big ], \end{equation}
\begin{equation} 0 = \mathcal{L}\tau (\eta) = \frac {1}{\varepsilon }\Big [\beta (u)\frac {\partial \tau }{\partial \eta }+\alpha (u)\frac {\partial ^2\tau }{\partial \eta ^2}\Big ], \end{equation}
which can be solved with the boundary condition (C11) to give
 \begin{equation} \tau (\eta) = C_0\left [1-\exp \left (-\frac {\beta }{\alpha }\eta \right)\right ]\!, \end{equation}
\begin{equation} \tau (\eta) = C_0\left [1-\exp \left (-\frac {\beta }{\alpha }\eta \right)\right ]\!, \end{equation}
where
 $C_0$
is some constant that we will determine next. One immediate observation is that the waiting time increases exponentially with
$C_0$
is some constant that we will determine next. One immediate observation is that the waiting time increases exponentially with
 $\eta$
, indicating the process spends most of its time near the boundary. Integrating the Kolmogorov equation
$\eta$
, indicating the process spends most of its time near the boundary. Integrating the Kolmogorov equation
 $\mathcal{L}\tau (x) = -\exp (-\Delta F/\varepsilon)$
against the invariant density
$\mathcal{L}\tau (x) = -\exp (-\Delta F/\varepsilon)$
against the invariant density
 $\rho _\infty (x)$
and applying the divergence theorem repeatedly, we get
$\rho _\infty (x)$
and applying the divergence theorem repeatedly, we get
 \begin{align} -\exp (-\Delta F/\varepsilon)\int _B\rho _\infty (x)\,{\rm d}x &= \int _B\mathcal{L}\tau (x)\rho _\infty (x) \,{\rm d}x \\[-12pt] \nonumber \end{align}
\begin{align} -\exp (-\Delta F/\varepsilon)\int _B\rho _\infty (x)\,{\rm d}x &= \int _B\mathcal{L}\tau (x)\rho _\infty (x) \,{\rm d}x \\[-12pt] \nonumber \end{align}
 \begin{align} &=\int _B b(x)\boldsymbol{\cdot }\boldsymbol{\nabla }\tau (x)\rho _\infty (x) + \frac {1}{2}\varepsilon a(x):\boldsymbol{\nabla }\boldsymbol{\nabla }\tau (x)\rho _\infty \,{\rm d}x \\[-12pt] \nonumber \end{align}
\begin{align} &=\int _B b(x)\boldsymbol{\cdot }\boldsymbol{\nabla }\tau (x)\rho _\infty (x) + \frac {1}{2}\varepsilon a(x):\boldsymbol{\nabla }\boldsymbol{\nabla }\tau (x)\rho _\infty \,{\rm d}x \\[-12pt] \nonumber \end{align}
This then leads to
 \begin{equation} C_0 = 2\exp (-\Delta F/\varepsilon)\dfrac { \int _B\rho _\infty (x)\, {\rm d}x}{ \int _{\partial D}\beta (u)\rho _\infty (u)\, {\rm d}u}, \end{equation}
\begin{equation} C_0 = 2\exp (-\Delta F/\varepsilon)\dfrac { \int _B\rho _\infty (x)\, {\rm d}x}{ \int _{\partial D}\beta (u)\rho _\infty (u)\, {\rm d}u}, \end{equation}
and the mean first passage time by (C9) is given by
 \begin{equation} w_B = 2\dfrac { \int _B\rho _\infty (x) \,{\rm d}x}{ \int _{\partial D}\beta (u)\rho _\infty (u)\, {\rm d}u}. \end{equation}
\begin{equation} w_B = 2\dfrac { \int _B\rho _\infty (x) \,{\rm d}x}{ \int _{\partial D}\beta (u)\rho _\infty (u)\, {\rm d}u}. \end{equation}
If the mobility operator has conserved quantity (Liu et al. Reference Liu, Sprittles and Grafke2024), the integrations must be performed over the hyperplane,
 \begin{equation} w_B = 2\dfrac { \int _{D/K}\rho _\infty (x)\, {\rm d}x}{ \int _{\partial D/K}\beta (u)\rho _\infty (u)\,{\rm d}u}, \end{equation}
\begin{equation} w_B = 2\dfrac { \int _{D/K}\rho _\infty (x)\, {\rm d}x}{ \int _{\partial D/K}\beta (u)\rho _\infty (u)\,{\rm d}u}, \end{equation}
where
 $K$
represents the dimensions perpendicular to the conserved quantities.
$K$
represents the dimensions perpendicular to the conserved quantities.
C.2. Average waiting time of adhesion for elastic membrane
We now apply the formula for the first passage time (C23) to the 1-D elastohydrodynamic thin film (2.6) with only bending and thermal noise on a periodic domain
 $x\in [0,1]$
. Its gradient flow form is given by
$x\in [0,1]$
. Its gradient flow form is given by
 \begin{equation} \frac {\partial h}{\partial t} = -m[h]\frac {\delta F}{\delta h} + \sqrt {2\varepsilon }m_{1/2}[h]\boldsymbol{\eta }, \end{equation}
\begin{equation} \frac {\partial h}{\partial t} = -m[h]\frac {\delta F}{\delta h} + \sqrt {2\varepsilon }m_{1/2}[h]\boldsymbol{\eta }, \end{equation}
where
 $F[h]$
is the energy functional
$F[h]$
is the energy functional
 \begin{equation} F[h] = \int _0^1 \frac {1}{2}\left (\frac {\partial ^2 h}{\partial x^2}\right)^2 \,{\rm d}x, \end{equation}
\begin{equation} F[h] = \int _0^1 \frac {1}{2}\left (\frac {\partial ^2 h}{\partial x^2}\right)^2 \,{\rm d}x, \end{equation}
 $\delta F/\delta h$
is the functional derivative of the energy functional, (for calculation of the functional derivative, see Sprittles et al. Reference Sprittles, Liu, Lockerby and Grafke2023; Liu et al. Reference Liu, Sprittles and Grafke2024)
$\delta F/\delta h$
is the functional derivative of the energy functional, (for calculation of the functional derivative, see Sprittles et al. Reference Sprittles, Liu, Lockerby and Grafke2023; Liu et al. Reference Liu, Sprittles and Grafke2024)
 \begin{equation} \frac {\delta F}{\delta h}[h] = \frac {\partial ^4 h}{\partial x^4}, \end{equation}
\begin{equation} \frac {\delta F}{\delta h}[h] = \frac {\partial ^4 h}{\partial x^4}, \end{equation}
 $m[h]$
is the mobility operator (acting on a test-function
$m[h]$
is the mobility operator (acting on a test-function
 $\xi$
)
$\xi$
)
 \begin{equation} m[h]\xi = -\frac {\partial }{\partial x}\left (h^3\frac {\partial }{\partial x}\xi \right)\!, \end{equation}
\begin{equation} m[h]\xi = -\frac {\partial }{\partial x}\left (h^3\frac {\partial }{\partial x}\xi \right)\!, \end{equation}
 $m_{1/2}[h]$
is the ‘square root’ of the mobility operator (acting on a test-function
$m_{1/2}[h]$
is the ‘square root’ of the mobility operator (acting on a test-function
 $\xi$
)
$\xi$
)
 \begin{equation} m_{1/2}[h]\xi = \sqrt {h^3}\frac {\partial }{\partial x}\xi , \end{equation}
\begin{equation} m_{1/2}[h]\xi = \sqrt {h^3}\frac {\partial }{\partial x}\xi , \end{equation}
 $\varepsilon$
is the noise amplitude
$\varepsilon$
is the noise amplitude
 \begin{equation} \varepsilon = Q_{\text{1-D}}^2/2 \end{equation}
\begin{equation} \varepsilon = Q_{\text{1-D}}^2/2 \end{equation}
and
 $\boldsymbol \eta$
is a Gaussian white noise. It is obvious that the mobility operator conserves mass given that a constant function is a zero eigenfunction (Liu et al. Reference Liu, Sprittles and Grafke2024) and the formula for the average waiting time must respect the conserved quantity, which is (C23). Without loss of generality, we let the mass to be
$\boldsymbol \eta$
is a Gaussian white noise. It is obvious that the mobility operator conserves mass given that a constant function is a zero eigenfunction (Liu et al. Reference Liu, Sprittles and Grafke2024) and the formula for the average waiting time must respect the conserved quantity, which is (C23). Without loss of generality, we let the mass to be
 $h_0$
and we would like to calculate the average time it takes for thermal fluctuations to drive a flat membrane
$h_0$
and we would like to calculate the average time it takes for thermal fluctuations to drive a flat membrane
 $h_A(x) = h_0$
to bend into a shape
$h_A(x) = h_0$
to bend into a shape
 $h(x)$
with minimum height
$h(x)$
with minimum height
 $h^*$
. This defines the boundary of the attractive basin
$h^*$
. This defines the boundary of the attractive basin
 $\partial D$
, namely, all the membrane shapes that have minimum height
$\partial D$
, namely, all the membrane shapes that have minimum height
 $h^*$
. Owing to the fact that the invariant distribution
$h^*$
. Owing to the fact that the invariant distribution
 $\rho _\infty$
is the Gibbs distribution, i.e. taking the form of an exponential, the integrals in (C23) can be approximated using the Laplace method
$\rho _\infty$
is the Gibbs distribution, i.e. taking the form of an exponential, the integrals in (C23) can be approximated using the Laplace method
 \begin{align} \int _{D/K}\rho _\infty \,{\rm d}h &= C\int _{D/K}\exp \left (-\frac {F[h]}{\varepsilon }\right) \,{\rm d}h \nonumber \\ &\approx C\exp \left (-\frac {F[h_0]}{\varepsilon }\right)\int _{D/K}\exp \left (-\frac {1}{2\varepsilon }(h-h_0)^TH[h_0](h-h_0)\right)\,{\rm d}h, \nonumber \\ \int _{\partial D/K}\beta (h)\rho _\infty \,{\rm d}h &= C\int _{\partial D/K}\beta (h)\exp \left (-\frac {F[h]}{\varepsilon }\right)\,{\rm d}h \nonumber \\ &\approx C\beta (h_B)\exp \left (-\frac {F[h_B]}{\varepsilon }\right)\int _{\partial D/K}\exp \left (-\frac {1}{2\varepsilon }(h-h_B)^T H[h_B](h-h_B)\right)\,{\rm d}h, \end{align}
\begin{align} \int _{D/K}\rho _\infty \,{\rm d}h &= C\int _{D/K}\exp \left (-\frac {F[h]}{\varepsilon }\right) \,{\rm d}h \nonumber \\ &\approx C\exp \left (-\frac {F[h_0]}{\varepsilon }\right)\int _{D/K}\exp \left (-\frac {1}{2\varepsilon }(h-h_0)^TH[h_0](h-h_0)\right)\,{\rm d}h, \nonumber \\ \int _{\partial D/K}\beta (h)\rho _\infty \,{\rm d}h &= C\int _{\partial D/K}\beta (h)\exp \left (-\frac {F[h]}{\varepsilon }\right)\,{\rm d}h \nonumber \\ &\approx C\beta (h_B)\exp \left (-\frac {F[h_B]}{\varepsilon }\right)\int _{\partial D/K}\exp \left (-\frac {1}{2\varepsilon }(h-h_B)^T H[h_B](h-h_B)\right)\,{\rm d}h, \end{align}
where
 $H[h] = \delta ^2 F/\delta h^2$
is the Hessian operator of the energy
$H[h] = \delta ^2 F/\delta h^2$
is the Hessian operator of the energy
 $F(h)$
, and
$F(h)$
, and
 \begin{equation} h^T H[h] h = \int _0^1 h\frac {\partial ^4 h}{\partial x^4}\, {\rm d}x \end{equation}
\begin{equation} h^T H[h] h = \int _0^1 h\frac {\partial ^4 h}{\partial x^4}\, {\rm d}x \end{equation}
can be interpreted as the inner product with respect to the Hessian (Liu et al. Reference Liu, Sprittles and Grafke2024). Apply these to (C23) and the average waiting time is given by
 \begin{equation} w_B = 2\frac {\int _{D/K}\exp \left (-\frac {1}{2\varepsilon }(h-h_0)^TH[h_0](h-h_0)\right)\,{\rm d}h}{\int _{\partial D/K}\exp \left (-\frac {1}{2\varepsilon }(h-h_B)^T H[h_B](h-h_B)\right)\,{\rm d}h}\exp \left (\frac {F[h_B]-F[h_0]}{\varepsilon }\right)\!, \end{equation}
\begin{equation} w_B = 2\frac {\int _{D/K}\exp \left (-\frac {1}{2\varepsilon }(h-h_0)^TH[h_0](h-h_0)\right)\,{\rm d}h}{\int _{\partial D/K}\exp \left (-\frac {1}{2\varepsilon }(h-h_B)^T H[h_B](h-h_B)\right)\,{\rm d}h}\exp \left (\frac {F[h_B]-F[h_0]}{\varepsilon }\right)\!, \end{equation}
whose form agrees with the result of the Large deviation theory (C9), and next, we need to determine the energy barrier
 $F[h_B]-F[h_0]$
and evaluate the integrals.
$F[h_B]-F[h_0]$
and evaluate the integrals.
C.2.1. Minimum energy profile for binding
If the noise amplitude is very small,
 $\varepsilon \ll 1$
, it is almost certain that the system will reach
$\varepsilon \ll 1$
, it is almost certain that the system will reach
 $\partial D$
while increasing the least energy possible. So finding the energy barrier
$\partial D$
while increasing the least energy possible. So finding the energy barrier
 $F[h_B]-F[h_0]$
is reduced to finding the
$F[h_B]-F[h_0]$
is reduced to finding the
 $h_B$
with minimum energy
$h_B$
with minimum energy
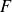 $F$
, which has a minimum height
$F$
, which has a minimum height
 $h^*$
, while also conserving mass and satisfying the boundary conditions of the problem. Mathematically, this corresponds to a constrained optimisation problem in which we seek to minimise the energy functional
$h^*$
, while also conserving mass and satisfying the boundary conditions of the problem. Mathematically, this corresponds to a constrained optimisation problem in which we seek to minimise the energy functional
 $F[h]$
. In this section, we use the Lagrange multiplier method (Boas Reference Boas2006) to find
$F[h]$
. In this section, we use the Lagrange multiplier method (Boas Reference Boas2006) to find
 $h_B(x)$
and its corresponding energy
$h_B(x)$
and its corresponding energy
 $F[h_B]$
. The Lagrangian taking into account the conservation of mass is
$F[h_B]$
. The Lagrangian taking into account the conservation of mass is
 \begin{equation} \tilde {F}=\frac {1}{2}\left (\frac {\partial \tilde {F}}{\partial x}\right)^2+\lambda (h-h_0), \end{equation}
\begin{equation} \tilde {F}=\frac {1}{2}\left (\frac {\partial \tilde {F}}{\partial x}\right)^2+\lambda (h-h_0), \end{equation}
where
 $\lambda$
is the Lagrange multiplier. The Euler–Lagrange equation is then given by
$\lambda$
is the Lagrange multiplier. The Euler–Lagrange equation is then given by
 \begin{equation} \lambda +\frac {\partial ^4 h}{\partial x^4}=0, \end{equation}
\begin{equation} \lambda +\frac {\partial ^4 h}{\partial x^4}=0, \end{equation}
for which the general solution is a fourth-order polynomial. In addition, the solution must satisfy our boundary and symmetry conditions, written as
 \begin{gather} h(0)=h(1)=h^* ,\end{gather}
\begin{gather} h(0)=h(1)=h^* ,\end{gather}
 \begin{gather} \frac {\partial h}{\partial x}\Bigr |_{x=1/2}=\frac {\partial ^3 h}{\partial x^3}\Bigr |_{x=1/2}=0 . \end{gather}
\begin{gather} \frac {\partial h}{\partial x}\Bigr |_{x=1/2}=\frac {\partial ^3 h}{\partial x^3}\Bigr |_{x=1/2}=0 . \end{gather}
The four boundary conditions in (C35) and (C36) are not linearly independent, so two more boundary conditions are required to determine the value of
 $\lambda$
and the other coefficients in the fourth-order polynomial. These are provided by requiring that the profile be smooth at the point of attachment (as it always is in our numerical simulations)
$\lambda$
and the other coefficients in the fourth-order polynomial. These are provided by requiring that the profile be smooth at the point of attachment (as it always is in our numerical simulations)
 \begin{gather} \frac {\partial h}{\partial x}\Bigr |_{x=0}=\frac {\partial h}{\partial x}\Bigr |_{x=1}=0 . \end{gather}
\begin{gather} \frac {\partial h}{\partial x}\Bigr |_{x=0}=\frac {\partial h}{\partial x}\Bigr |_{x=1}=0 . \end{gather}
It is interesting to note that a smooth profile is required for a bending-dominated membrane, when this is not the case for tension-dominated films (Sprittles et al. Reference Sprittles, Liu, Lockerby and Grafke2023). A physical interpretation for this is that the curvature, and thus bending energy, goes to infinity at a discontinuity in
 $\partial h/\partial x$
. This is also reflected in the smooth equilibrium profile for a bending-dominated blister as opposed to the sharp edge (at least macroscopically) for a capillary droplet.
$\partial h/\partial x$
. This is also reflected in the smooth equilibrium profile for a bending-dominated blister as opposed to the sharp edge (at least macroscopically) for a capillary droplet.
When the boundary conditions of (C35)–(C37) and conservation of mass are applied to a fourth-order polynomial, we obtain the following profile:
 \begin{equation} h_B(x)=h^*+30(h_0-h^*)(x^4-2x^3+x^2). \end{equation}
\begin{equation} h_B(x)=h^*+30(h_0-h^*)(x^4-2x^3+x^2). \end{equation}
This is the profile we predict as the average profile at the moment of binding when the parameters
 $Q_{\text{1-D}}$
and
$Q_{\text{1-D}}$
and
 $h_0-h^*$
are selected such that the binding event is sufficiently rare. This prediction is confirmed by the data presented in figure 2, where the dashed blue line represents (C38) and the black dots represent the average profile for 15 individual simulations.
$h_0-h^*$
are selected such that the binding event is sufficiently rare. This prediction is confirmed by the data presented in figure 2, where the dashed blue line represents (C38) and the black dots represent the average profile for 15 individual simulations.
The energy barrier is then given by computing
 $F$
for the profile of (C38),
$F$
for the profile of (C38),
 \begin{equation} F[h_B]-F[h_0]= 360 (h_0-h^*)^2. \end{equation}
\begin{equation} F[h_B]-F[h_0]= 360 (h_0-h^*)^2. \end{equation}
C.2.2. Evaluation of the integrals
We now turn to the integrals in (C32). Due to periodicity, membrane shapes can be decomposed into Fourier modes. At the cost of a small error, this allows us to evaluate the integrals analytically. Decomposing
 $h-h_0$
into Fourier modes gives
$h-h_0$
into Fourier modes gives
 \begin{equation} h(x)-h_0 = \sum _{n=1}^\infty a_n\cos (2\pi nx) + b_n\sin (2\pi nx) \end{equation}
\begin{equation} h(x)-h_0 = \sum _{n=1}^\infty a_n\cos (2\pi nx) + b_n\sin (2\pi nx) \end{equation}
and so
 \begin{equation} (h-h_0)^T H[h_0](h-h_0) = \sum _{n=1}^\infty (2\pi n)^8\frac {1}{2}\big(a_n^2+b_n^2\big), \end{equation}
\begin{equation} (h-h_0)^T H[h_0](h-h_0) = \sum _{n=1}^\infty (2\pi n)^8\frac {1}{2}\big(a_n^2+b_n^2\big), \end{equation}
which gives us
 \begin{align} \notag &\int _{D/K}\exp \left (-\frac {1}{2\varepsilon }(h-h_0)^TH[h_0](h-h_0)\right)\,{\rm d}h \\ \notag &\qquad =\int _{-\infty }^\infty \exp \left (-\frac {1}{2\varepsilon }\sum _{n=1}^{\infty }(2\pi n)^8\frac {1}{2}(a_n^2+b_n^2)\right) \prod _{p=1}^\infty {\rm d}a_n {\rm d}b_n\\ &\qquad =\prod _{n=1}^\infty \frac {4\varepsilon \pi }{(2\pi n)^8}. \end{align}
\begin{align} \notag &\int _{D/K}\exp \left (-\frac {1}{2\varepsilon }(h-h_0)^TH[h_0](h-h_0)\right)\,{\rm d}h \\ \notag &\qquad =\int _{-\infty }^\infty \exp \left (-\frac {1}{2\varepsilon }\sum _{n=1}^{\infty }(2\pi n)^8\frac {1}{2}(a_n^2+b_n^2)\right) \prod _{p=1}^\infty {\rm d}a_n {\rm d}b_n\\ &\qquad =\prod _{n=1}^\infty \frac {4\varepsilon \pi }{(2\pi n)^8}. \end{align}
We can also decompose
 $h-h_B$
into Fourier modes, however, care must be taken when integrating over
$h-h_B$
into Fourier modes, however, care must be taken when integrating over
 $\partial D/K$
by retaining only the modes parallel to
$\partial D/K$
by retaining only the modes parallel to
 $\partial D$
at
$\partial D$
at
 $h_B$
, or equivalently, by removing the modes perpendicular to
$h_B$
, or equivalently, by removing the modes perpendicular to
 $\partial D$
at
$\partial D$
at
 $h_B$
. Due to the way we constructed
$h_B$
. Due to the way we constructed
 $\partial D$
, the only mode to remove is
$\partial D$
, the only mode to remove is
 $\hat n(h_B)$
, that is,
$\hat n(h_B)$
, that is,
 \begin{equation} \hat n(h_B) = \frac {\delta F[h_B]/\delta h}{|\delta F[h_B]/\delta h|} = 2\cos (2\pi x). \end{equation}
\begin{equation} \hat n(h_B) = \frac {\delta F[h_B]/\delta h}{|\delta F[h_B]/\delta h|} = 2\cos (2\pi x). \end{equation}
Therefore, the decomposition is given by
 \begin{equation} h(x)-h_B(x) = d_1\sin (2\pi x) + \sum _{n=2}^\infty c_n\cos (2\pi nx)+d_n\sin (2\pi nx), \end{equation}
\begin{equation} h(x)-h_B(x) = d_1\sin (2\pi x) + \sum _{n=2}^\infty c_n\cos (2\pi nx)+d_n\sin (2\pi nx), \end{equation}
which leads to
 \begin{equation} (h-h_B)^T H[h_B](h-h_B) = \frac {1}{2}(2\pi)^8 d_1^2 + \sum _{n=2}^\infty (2\pi n)^8\frac {1}{2}\big(a_n^2+b_n^2\big) \end{equation}
\begin{equation} (h-h_B)^T H[h_B](h-h_B) = \frac {1}{2}(2\pi)^8 d_1^2 + \sum _{n=2}^\infty (2\pi n)^8\frac {1}{2}\big(a_n^2+b_n^2\big) \end{equation}
and so
 \begin{align} \notag &\int _{\partial D/K}\exp \left (-\frac {1}{2\varepsilon }(h-h_B)^TH[h_B](h-h_B)\right)\,{\rm d}h \\ \notag =& \int _{-\infty }^\infty \exp \left (-\frac {1}{2\varepsilon }(2\pi)^8\frac {1}{2}d_1^2\right)\, {\rm d}d_1\int _{-\infty }^\infty \exp \left (-\frac {1}{2\varepsilon }\sum _{n=2}^\infty (2\pi n)^8\frac {1}{2}(c_n^2+d_n^2)\right)\prod _{n=2}^\infty {\rm d}c_n \,{\rm d}d_n\\ =& \sqrt {\frac {2\varepsilon \pi }{(2\pi)^8}}\prod _{n=2}^\infty \frac {4\varepsilon \pi }{(2\pi n)^8}. \end{align}
\begin{align} \notag &\int _{\partial D/K}\exp \left (-\frac {1}{2\varepsilon }(h-h_B)^TH[h_B](h-h_B)\right)\,{\rm d}h \\ \notag =& \int _{-\infty }^\infty \exp \left (-\frac {1}{2\varepsilon }(2\pi)^8\frac {1}{2}d_1^2\right)\, {\rm d}d_1\int _{-\infty }^\infty \exp \left (-\frac {1}{2\varepsilon }\sum _{n=2}^\infty (2\pi n)^8\frac {1}{2}(c_n^2+d_n^2)\right)\prod _{n=2}^\infty {\rm d}c_n \,{\rm d}d_n\\ =& \sqrt {\frac {2\varepsilon \pi }{(2\pi)^8}}\prod _{n=2}^\infty \frac {4\varepsilon \pi }{(2\pi n)^8}. \end{align}
C.2.3. Final result
Combining (C23) (C42) (C46), we get
 \begin{equation} w_B = \frac {2}{\beta (h_B)}\exp \left (\frac {F[h_B]-F[h_0]}{\varepsilon }\right)\frac {4\varepsilon \pi }{(2\pi)^8}, \end{equation}
\begin{equation} w_B = \frac {2}{\beta (h_B)}\exp \left (\frac {F[h_B]-F[h_0]}{\varepsilon }\right)\frac {4\varepsilon \pi }{(2\pi)^8}, \end{equation}
and the only missing part is
 $\beta (h_B)$
. By (C15) (C27) (C26), we have
$\beta (h_B)$
. By (C15) (C27) (C26), we have
 \begin{align} \beta (h_B) &= -\hat n(h_B)\boldsymbol{\cdot }\left (M(h_B)\frac {\delta F}{\delta h}(h_B)\right)=-(h-h^*)(2\pi)^6\left (\frac {1}{2}h_0^3+\frac {3}{8}h_0(h_0-h^*)^2\right)\!, \end{align}
\begin{align} \beta (h_B) &= -\hat n(h_B)\boldsymbol{\cdot }\left (M(h_B)\frac {\delta F}{\delta h}(h_B)\right)=-(h-h^*)(2\pi)^6\left (\frac {1}{2}h_0^3+\frac {3}{8}h_0(h_0-h^*)^2\right)\!, \end{align}
where the inner product is interpreted as integral, same as (C31). Finally, with (C25), we arrive at the expression for the average waiting time used in the main text (3.2),
 \begin{equation} \langle t_B \rangle = w_B = \frac {1}{\beta (h_B)}\sqrt {\frac {Q_\textrm{1-D}^2}{(2\pi)^7}} \exp \left (720\left (\frac {h_0-h^*}{Q_\textrm{1-D}}\right)^2\right)\!. \end{equation}
\begin{equation} \langle t_B \rangle = w_B = \frac {1}{\beta (h_B)}\sqrt {\frac {Q_\textrm{1-D}^2}{(2\pi)^7}} \exp \left (720\left (\frac {h_0-h^*}{Q_\textrm{1-D}}\right)^2\right)\!. \end{equation}
Appendix D. Domain coarsening theory
In § 4, we present the results of simulations in which lumens are formed in the space between two membranes, and then coarsen with time. Here, we provide more details about how we quantify and rationalise the coarsening behaviour. First, we will discuss how we compute the characteristic length scale
 $L_c$
in light of previous work on phase separating systems. Second, we will provide more details on the theoretical description from § 4.1 which we use to rationalise the decreased growth rate for bending-driven coarsening.
$L_c$
in light of previous work on phase separating systems. Second, we will provide more details on the theoretical description from § 4.1 which we use to rationalise the decreased growth rate for bending-driven coarsening.
D.1.
Computation of the scaling length
 $L_c$
$L_c$

The separation of two phases in a 2-D domain is always a complex process, where the fluxes giving rise to coarsening are inherently local phenomena that depend on the specific morphology of the profile. For non-uniformly distributed profiles such as those shown in figure 1(a,b), it is not straightforward to describe the morphology of the profile. Nevertheless, the system does exhibit a clear and obvious change with respect to time, as shown in figures 3 and 5. To gain a quantitative understanding of these phenomena, a statistical approach can be used to gain insight into the ensemble-averaged behaviour of such profiles over time. Specifically, the well-established scaling hypothesis for phase separating dynamical systems states that during the late-stage of coarsening, the domain structure is self-similar with respect to time when the length is rescaled by a single length scale
 $L_c(t)$
(Furukawa Reference Furukawa1985; Bray Reference Bray1994). When systems demonstrate this type of behaviour, the morphologies of individual realisations of the system are still distinct, but multiple realisations will look similar to the eye at a single time step. If one then zooms out so that the length scale increases in accordance with
$L_c(t)$
(Furukawa Reference Furukawa1985; Bray Reference Bray1994). When systems demonstrate this type of behaviour, the morphologies of individual realisations of the system are still distinct, but multiple realisations will look similar to the eye at a single time step. If one then zooms out so that the length scale increases in accordance with
 $L_c(t)$
, the morphologies of multiple trajectories will be indistinguishable even as time increases (Camley & Brown Reference Camley and Brown2011). Many systems have been shown to demonstrate this type of behaviour both numerically and experimentally (Komura et al. Reference Komura, Osamura, Fujii and Takeda1985; Sung et al. Reference Sung, Karim, Douglas and Han1996; Furukawa Reference Furukawa2000; Livet et al. Reference Livet, Bley, Caudron, Geissler, Abernathy, Detlefs, Grübel and Sutton2001; Lal et al. Reference Lal, Lurio, Liang, Narayanan, Darling and Sutton2020; Tateno & Tanaka Reference Tateno and Tanaka2021; Su et al. Reference Su, Ho, Gettel, Rowland, Keating and Parikh2024).
$L_c(t)$
, the morphologies of multiple trajectories will be indistinguishable even as time increases (Camley & Brown Reference Camley and Brown2011). Many systems have been shown to demonstrate this type of behaviour both numerically and experimentally (Komura et al. Reference Komura, Osamura, Fujii and Takeda1985; Sung et al. Reference Sung, Karim, Douglas and Han1996; Furukawa Reference Furukawa2000; Livet et al. Reference Livet, Bley, Caudron, Geissler, Abernathy, Detlefs, Grübel and Sutton2001; Lal et al. Reference Lal, Lurio, Liang, Narayanan, Darling and Sutton2020; Tateno & Tanaka Reference Tateno and Tanaka2021; Su et al. Reference Su, Ho, Gettel, Rowland, Keating and Parikh2024).
The scaling hypothesis is thus a powerful concept that allows us to meaningfully understand coarsening domains. The question that remains, however, is how the length scale
 $L_c(t)$
can be calculated from a height profile
$L_c(t)$
can be calculated from a height profile
 $h(x,y,t)$
. The scaling hypothesis suggests that the domain structure should be independent of time except for a dependence on
$h(x,y,t)$
. The scaling hypothesis suggests that the domain structure should be independent of time except for a dependence on
 $L_c(t)$
. The structure can by represented by its equal-time correlation function, which is defined as
$L_c(t)$
. The structure can by represented by its equal-time correlation function, which is defined as
 \begin{equation} C(\boldsymbol{r},t)=\langle h(\boldsymbol{x}+\boldsymbol{r},t)h(\boldsymbol{x},t)\rangle , \end{equation}
\begin{equation} C(\boldsymbol{r},t)=\langle h(\boldsymbol{x}+\boldsymbol{r},t)h(\boldsymbol{x},t)\rangle , \end{equation}
where
 $\boldsymbol{x}$
is the position vector
$\boldsymbol{x}$
is the position vector
 $(x,y)$
,
$(x,y)$
,
 $\boldsymbol{r}$
is a displacement vector and the angular brackets represent an ensemble average. If the scaling hypothesis is valid,
$\boldsymbol{r}$
is a displacement vector and the angular brackets represent an ensemble average. If the scaling hypothesis is valid,
 $C(\boldsymbol{r},t)$
should behave according to
$C(\boldsymbol{r},t)$
should behave according to
 \begin{equation} C(\boldsymbol{r},t)=f\left (\frac {r}{L_c(t)}\right)\!. \end{equation}
\begin{equation} C(\boldsymbol{r},t)=f\left (\frac {r}{L_c(t)}\right)\!. \end{equation}
The equal-time structure factor,
 $S(\boldsymbol {k},t)$
, is defined as
$S(\boldsymbol {k},t)$
, is defined as
 \begin{equation} S(\boldsymbol {k},t)=\langle h_{\boldsymbol {k}}(t)h_{\boldsymbol {-k}}(t)\rangle , \end{equation}
\begin{equation} S(\boldsymbol {k},t)=\langle h_{\boldsymbol {k}}(t)h_{\boldsymbol {-k}}(t)\rangle , \end{equation}
where
 $\boldsymbol{k}$
is now a wavevector and
$\boldsymbol{k}$
is now a wavevector and
 $h_k$
is the 2-D Fourier transform of
$h_k$
is the 2-D Fourier transform of
 $h(\boldsymbol{x)}$
. Since
$h(\boldsymbol{x)}$
. Since
 $S(\boldsymbol {k},t)$
is simply the Fourier transform of
$S(\boldsymbol {k},t)$
is simply the Fourier transform of
 $C(\boldsymbol{r},t)$
, it must have the scaling form
$C(\boldsymbol{r},t)$
, it must have the scaling form
 \begin{equation} S(\boldsymbol {k},t)=L_c^2g(k,L_c), \end{equation}
\begin{equation} S(\boldsymbol {k},t)=L_c^2g(k,L_c), \end{equation}
where
 $g$
is the Fourier transform of
$g$
is the Fourier transform of
 $f$
(Bray Reference Bray1994). To calculate
$f$
(Bray Reference Bray1994). To calculate
 $L_c$
one needs to extract a length scale from
$L_c$
one needs to extract a length scale from
 $S(\boldsymbol {k},t)$
. This is commonly done by taking a moment of spherically averaged structure function
$S(\boldsymbol {k},t)$
. This is commonly done by taking a moment of spherically averaged structure function
 $S(k,t)$
(Shinozaki & Oono Reference Shinozaki and Oono1993; Furukawa Reference Furukawa2000; Camley & Brown Reference Camley and Brown2011). In this paper, we found more consistent results by calculating the characteristic length using the following expression as suggested by Shinozaki & Oono (Reference Shinozaki and Oono1993):
$S(k,t)$
(Shinozaki & Oono Reference Shinozaki and Oono1993; Furukawa Reference Furukawa2000; Camley & Brown Reference Camley and Brown2011). In this paper, we found more consistent results by calculating the characteristic length using the following expression as suggested by Shinozaki & Oono (Reference Shinozaki and Oono1993):
 \begin{equation} L_c =2\pi \frac {\sum \limits _{k\ne 0} |\boldsymbol{k}|^{-2}S(\boldsymbol{k})}{\sum \limits _{k\ne 0} |\boldsymbol{k}|^{-1}S(\boldsymbol{k})}. \end{equation}
\begin{equation} L_c =2\pi \frac {\sum \limits _{k\ne 0} |\boldsymbol{k}|^{-2}S(\boldsymbol{k})}{\sum \limits _{k\ne 0} |\boldsymbol{k}|^{-1}S(\boldsymbol{k})}. \end{equation}
Our procedure for calculating
 $L_c$
thus consisted of the following. First,
$L_c$
thus consisted of the following. First,
 $h_{\boldsymbol {k}}$
was computed for an individual profile by taking a 2-D fast Fourier transform of the height profile
$h_{\boldsymbol {k}}$
was computed for an individual profile by taking a 2-D fast Fourier transform of the height profile
 $h(x,y)$
. Next,
$h(x,y)$
. Next,
 $h_{\boldsymbol {k}}(t)h_{\boldsymbol {-k}}$
was computed for each profile, since it is equal to the Fourier transform of
$h_{\boldsymbol {k}}(t)h_{\boldsymbol {-k}}$
was computed for each profile, since it is equal to the Fourier transform of
 $C(\boldsymbol {r})$
. Then,
$C(\boldsymbol {r})$
. Then,
 $S(\boldsymbol {k})$
is computed as the ensemble average of
$S(\boldsymbol {k})$
is computed as the ensemble average of
 $h_{\boldsymbol {k}}h_{\boldsymbol {-k}}$
. Finally, (D5) is used to compute
$h_{\boldsymbol {k}}h_{\boldsymbol {-k}}$
. Finally, (D5) is used to compute
 $L_c$
.
$L_c$
.
D.2. Coarsening rate for bending-driven coarsening
To predict how the coarsening rate will change when the tension term is replaced by the bending term in (2.3), we follow the scaling logic of Bray (Reference Bray1994). We note that the following theory only takes into account the change of the pressure term. It does not account for the effects of nonlinear mobility or a complicated single-well potential, which are included in our mathematical model. The simplified system we study in this section is thus the following:
 \begin{equation} \frac {\partial h}{\partial t}= {\nabla} ^2 \frac {\delta F }{\delta h}, \end{equation}
\begin{equation} \frac {\partial h}{\partial t}= {\nabla} ^2 \frac {\delta F }{\delta h}, \end{equation}
where the free energy
 $F$
is given by
$F$
is given by
 \begin{equation} F=\int \left (\frac {1}{2}\big ({\nabla} ^2h\big)^2+V(h)\right)\!. \end{equation}
\begin{equation} F=\int \left (\frac {1}{2}\big ({\nabla} ^2h\big)^2+V(h)\right)\!. \end{equation}
Our goal will be to find out the motion of domain walls between two bulk regions, as depicted in figure 9. We consider
 $V(h)$
to be a symmetric double-well potential with wells of even depth located at
$V(h)$
to be a symmetric double-well potential with wells of even depth located at
 $h=1$
and
$h=1$
and
 $h=1+\Delta h$
. Although this obviously does not match the energy from protein binding described in (A4), we expect the scaling rule to be insensitive to the exact form of
$h=1+\Delta h$
. Although this obviously does not match the energy from protein binding described in (A4), we expect the scaling rule to be insensitive to the exact form of
 $V(h)$
. This is indeed justified by the numerical results in § 4.2, which validate the
$V(h)$
. This is indeed justified by the numerical results in § 4.2, which validate the
 $1/3$
power law predicted by Bray for constant mobility films even when the single-well potential of (A4) is used.
$1/3$
power law predicted by Bray for constant mobility films even when the single-well potential of (A4) is used.

Figure 9. Schematic showing the physical picture of ‘diffusive’ domain coarsening. Two bulk phases are separated by a curved interface. The unit vector
 $\boldsymbol{\hat {g}}$
points in the direction normal to the interface.
$\boldsymbol{\hat {g}}$
points in the direction normal to the interface.
When the energy from (D7) is inserted into (D6), we get
 \begin{equation} \frac {\partial h}{\partial t} = {\nabla} ^2\big ({\nabla} ^4h+ V^{\prime}(h)\big). \end{equation}
\begin{equation} \frac {\partial h}{\partial t} = {\nabla} ^2\big ({\nabla} ^4h+ V^{\prime}(h)\big). \end{equation}
We start by investigating the bulk phases where
 $h$
is in the bound state and thus only slightly deviates from its equilibrium value. We linearise (D8) by introducing
$h$
is in the bound state and thus only slightly deviates from its equilibrium value. We linearise (D8) by introducing
 $h=1+\tilde {h}$
. This gives us the following linearised equation:
$h=1+\tilde {h}$
. This gives us the following linearised equation:
 \begin{equation} \frac {\partial \tilde {h}}{\partial t} = {\nabla} ^6\tilde {h}+ V^{\prime\prime}(1){\nabla} ^2\tilde {h} . \end{equation}
\begin{equation} \frac {\partial \tilde {h}}{\partial t} = {\nabla} ^6\tilde {h}+ V^{\prime\prime}(1){\nabla} ^2\tilde {h} . \end{equation}
Since the characteristic domain size
 $L_c$
is large during late-stage coarsening, the
$L_c$
is large during late-stage coarsening, the
 ${\sim} {\nabla} ^6$
term can be neglected, which reduces (D9) to a diffusion equation for
${\sim} {\nabla} ^6$
term can be neglected, which reduces (D9) to a diffusion equation for
 $\tilde {h}$
with diffusion coefficient
$\tilde {h}$
with diffusion coefficient
 $V^{\prime\prime}(1)$
. Now, we expect that during late-stage coarsening, the diffusion field relaxes much faster than the motion of the domain walls. We can thus assume that this diffusion field is always in a quasi-equilibrium with the location of the interface, i.e.
$V^{\prime\prime}(1)$
. Now, we expect that during late-stage coarsening, the diffusion field relaxes much faster than the motion of the domain walls. We can thus assume that this diffusion field is always in a quasi-equilibrium with the location of the interface, i.e.
 \begin{equation} {\nabla} ^2h=0 \end{equation}
\begin{equation} {\nabla} ^2h=0 \end{equation}
in the bulk regions away from the interface.
Before we start investigating the interfaces between the bulk phases, we will reformulate (D8) in terms of a flux
 $\boldsymbol{j}$
given by the gradient of a chemical potential
$\boldsymbol{j}$
given by the gradient of a chemical potential
 $\varPhi$
(which is equivalent to the pressure in our system):
$\varPhi$
(which is equivalent to the pressure in our system):
 \begin{align} \frac {\partial h}{\partial t}&= -\boldsymbol{\nabla }\boldsymbol{\cdot }\boldsymbol{j}, \end{align}
\begin{align} \frac {\partial h}{\partial t}&= -\boldsymbol{\nabla }\boldsymbol{\cdot }\boldsymbol{j}, \end{align}
 \begin{align} \boldsymbol{j}&=-\boldsymbol{\nabla }\varPhi, \end{align}
\begin{align} \boldsymbol{j}&=-\boldsymbol{\nabla }\varPhi, \end{align}
 \begin{align} \varPhi &= \frac {\partial V}{\partial h} +{\nabla} ^4h . \end{align}
\begin{align} \varPhi &= \frac {\partial V}{\partial h} +{\nabla} ^4h . \end{align}
If we now introduce the linearised
 $h=1+\tilde {h}$
into (D13), we get
$h=1+\tilde {h}$
into (D13), we get
 \begin{equation} \varPhi = {\nabla} ^4\tilde {h}+ V^{\prime\prime}(1)\tilde {h} . \end{equation}
\begin{equation} \varPhi = {\nabla} ^4\tilde {h}+ V^{\prime\prime}(1)\tilde {h} . \end{equation}
Again, during the latter stages of coarsening, the length scale is large, meaning that the
 ${\sim} {\nabla} ^4$
term in (D14) is negligible. This gives us
${\sim} {\nabla} ^4$
term in (D14) is negligible. This gives us
 $\varPhi \sim \tilde {h}$
in the bulk phases, meaning that
$\varPhi \sim \tilde {h}$
in the bulk phases, meaning that
 $\varPhi$
also satisfies the Laplace equation
$\varPhi$
also satisfies the Laplace equation
 \begin{equation}{\nabla} ^2\varPhi =0. \end{equation}
\begin{equation}{\nabla} ^2\varPhi =0. \end{equation}
To find out what
 $\varPhi$
is at the boundary between the bulk phases, we first introduce the alternate coordinate system shown in figure 9 to simplify the vector calculus calculations. The unit vector
$\varPhi$
is at the boundary between the bulk phases, we first introduce the alternate coordinate system shown in figure 9 to simplify the vector calculus calculations. The unit vector
 $\boldsymbol{\hat {g}}$
points in the direction perpendicular to the interface (
$\boldsymbol{\hat {g}}$
points in the direction perpendicular to the interface (
 $g=\pm \infty$
in the bulk and
$g=\pm \infty$
in the bulk and
 $g=0$
at the centre of the interface). In the following, we will study an interface with the circular geometry shown in figure 9, meaning that
$g=0$
at the centre of the interface). In the following, we will study an interface with the circular geometry shown in figure 9, meaning that
 $\boldsymbol{\nabla }\phi = (\partial \phi /\partial g)\boldsymbol{\hat {g}}$
and
$\boldsymbol{\nabla }\phi = (\partial \phi /\partial g)\boldsymbol{\hat {g}}$
and
 $\boldsymbol{\nabla }\boldsymbol{\cdot }\boldsymbol{\hat {g}}=1/R$
near the interface. This allows us to compute the Laplacian as
$\boldsymbol{\nabla }\boldsymbol{\cdot }\boldsymbol{\hat {g}}=1/R$
near the interface. This allows us to compute the Laplacian as
 ${\nabla} ^2\phi =\partial ^2\phi /\partial g^2 + (\partial \phi /\partial g) \boldsymbol{\nabla }\boldsymbol{\cdot }\boldsymbol{\hat {g}}$
. When these identities are inserted into (D13), we find that
${\nabla} ^2\phi =\partial ^2\phi /\partial g^2 + (\partial \phi /\partial g) \boldsymbol{\nabla }\boldsymbol{\cdot }\boldsymbol{\hat {g}}$
. When these identities are inserted into (D13), we find that
 \begin{equation} \varPhi = \frac {\partial V}{\partial h} +\frac {\partial ^4 h}{\partial g^4}+\frac {2}{R}\frac {\partial ^3 h}{\partial g^3}-\frac {1}{R^2}\frac {\partial ^2 h}{\partial g^2}+\frac {1}{R^3}\frac {\partial h}{\partial g}. \end{equation}
\begin{equation} \varPhi = \frac {\partial V}{\partial h} +\frac {\partial ^4 h}{\partial g^4}+\frac {2}{R}\frac {\partial ^3 h}{\partial g^3}-\frac {1}{R^2}\frac {\partial ^2 h}{\partial g^2}+\frac {1}{R^3}\frac {\partial h}{\partial g}. \end{equation}
To find the value of
 $\varPhi$
at an interface with radius of curvature
$\varPhi$
at an interface with radius of curvature
 $R$
, we then multiply (D16) by
$R$
, we then multiply (D16) by
 $\partial h/\partial g$
and integrate across the interface from
$\partial h/\partial g$
and integrate across the interface from
 $g=-\infty$
to
$g=-\infty$
to
 $g=\infty$
. This gives us
$g=\infty$
. This gives us
 \begin{align} \notag\varPhi \Delta h &= \Delta V +\int ^\infty _{-\infty }\frac {\partial ^4 h}{\partial g^4}\frac {\partial h}{\partial g}\,{\rm d}g +\frac {2}{R}\int ^\infty _{-\infty }\frac {\partial ^3 h}{\partial g^3}\frac {\partial h}{\partial g}\,{\rm d}g \\ &\quad-\frac {1}{2R^2}\int ^\infty _{-\infty }\frac {\partial }{\partial g}\left (\frac {\partial h}{\partial g}\right)^2\,{\rm d}g +\frac {1}{R^3}\int ^\infty _{-\infty }\left (\frac {\partial h}{\partial g}\right)^2\,{\rm d}g. \end{align}
\begin{align} \notag\varPhi \Delta h &= \Delta V +\int ^\infty _{-\infty }\frac {\partial ^4 h}{\partial g^4}\frac {\partial h}{\partial g}\,{\rm d}g +\frac {2}{R}\int ^\infty _{-\infty }\frac {\partial ^3 h}{\partial g^3}\frac {\partial h}{\partial g}\,{\rm d}g \\ &\quad-\frac {1}{2R^2}\int ^\infty _{-\infty }\frac {\partial }{\partial g}\left (\frac {\partial h}{\partial g}\right)^2\,{\rm d}g +\frac {1}{R^3}\int ^\infty _{-\infty }\left (\frac {\partial h}{\partial g}\right)^2\,{\rm d}g. \end{align}
The
 $\Delta V$
term on the right-hand side of (D17) is zero because the potential wells we are considering have equal depth. The third term on the right-hand side disappears to to the bulk boundary condition
$\Delta V$
term on the right-hand side of (D17) is zero because the potential wells we are considering have equal depth. The third term on the right-hand side disappears to to the bulk boundary condition
 ${\partial h}/{\partial g}\Big \rvert _{\pm \infty }=0$
. The second term on the right-hand side disappears upon using integration by parts and implementing the boundary conditions
${\partial h}/{\partial g}\Big \rvert _{\pm \infty }=0$
. The second term on the right-hand side disappears upon using integration by parts and implementing the boundary conditions
 ${\partial h}/{\partial g}\Big \rvert _{\pm \infty }={\partial ^2 h}/{\partial g^2}\Big \rvert _{\pm \infty }=0$
. Equation (D17) then reduces to
${\partial h}/{\partial g}\Big \rvert _{\pm \infty }={\partial ^2 h}/{\partial g^2}\Big \rvert _{\pm \infty }=0$
. Equation (D17) then reduces to
 \begin{equation} \varPhi \Delta h = -\frac {2}{R}\int ^\infty _{-\infty }\left (\frac {\partial ^2 h}{\partial g^2}\right)^2\,{\rm d}g+\frac {1}{R^3}\int ^\infty _{-\infty }\left (\frac {\partial h}{\partial g}\right)^2\,{\rm d}g, \end{equation}
\begin{equation} \varPhi \Delta h = -\frac {2}{R}\int ^\infty _{-\infty }\left (\frac {\partial ^2 h}{\partial g^2}\right)^2\,{\rm d}g+\frac {1}{R^3}\int ^\infty _{-\infty }\left (\frac {\partial h}{\partial g}\right)^2\,{\rm d}g, \end{equation}
which is the equivalent of the Gibbs–Thompson boundary condition for an interface with bending energy. The factors
 $-2\int ^\infty _{-\infty} ( {\partial ^2 h}/{\partial g^2})^2\,{\rm d}g$
and
$-2\int ^\infty _{-\infty} ( {\partial ^2 h}/{\partial g^2})^2\,{\rm d}g$
and
 $\int ^\infty _{-\infty} ( {\partial h}/{\partial g})^2\,{\rm d}g$
can be interpreted as effective ‘line tension’ coefficients
$\int ^\infty _{-\infty} ( {\partial h}/{\partial g})^2\,{\rm d}g$
can be interpreted as effective ‘line tension’ coefficients
 $\varGamma _1$
and
$\varGamma _1$
and
 $\varGamma _2$
, respectively, as they represent an energy per unit length associated with the circular interface between the two phases. Equation (D18) can thus be rewritten as
$\varGamma _2$
, respectively, as they represent an energy per unit length associated with the circular interface between the two phases. Equation (D18) can thus be rewritten as
 \begin{equation} \varPhi \Delta h = \frac {\varGamma _1}{R}+\frac {\varGamma _2}{R^3}. \end{equation}
\begin{equation} \varPhi \Delta h = \frac {\varGamma _1}{R}+\frac {\varGamma _2}{R^3}. \end{equation}
Having found the chemical potential of a curved interface, we can now use (D12) to find the flux
 $\boldsymbol{j}$
. The velocity
$\boldsymbol{j}$
. The velocity
 $v_{int}$
at which the interface moves can be found from the difference in flux leaving the interface and flux entering the interface:
$v_{int}$
at which the interface moves can be found from the difference in flux leaving the interface and flux entering the interface:
 \begin{equation} v_{int}\Delta h =\boldsymbol {j}_{\textit{out}}-\boldsymbol {j}_{\textit{in}}=-\left (\left (\frac {\partial \varPhi }{\partial g}\right)_{R+\epsilon }-\left (\frac {\partial \varPhi }{\partial g}\right)_{R-\epsilon }\right)\!. \end{equation}
\begin{equation} v_{int}\Delta h =\boldsymbol {j}_{\textit{out}}-\boldsymbol {j}_{\textit{in}}=-\left (\left (\frac {\partial \varPhi }{\partial g}\right)_{R+\epsilon }-\left (\frac {\partial \varPhi }{\partial g}\right)_{R-\epsilon }\right)\!. \end{equation}
Setting
 $v_{int}$
equal to the rate of the change of the characteristic length scale
$v_{int}$
equal to the rate of the change of the characteristic length scale
 $L_c$
(in this case, the radius
$L_c$
(in this case, the radius
 $R$
) and using the chemical potential from (D19), we can get a prediction for how
$R$
) and using the chemical potential from (D19), we can get a prediction for how
 $L_c$
will grow with time:
$L_c$
will grow with time:
 \begin{equation} \frac {{\rm d}L_c}{{\rm d}t}\sim \frac {\varGamma _1}{L_c^2}+\frac {\varGamma _2}{L_c^4}. \end{equation}
\begin{equation} \frac {{\rm d}L_c}{{\rm d}t}\sim \frac {\varGamma _1}{L_c^2}+\frac {\varGamma _2}{L_c^4}. \end{equation}
This growth leads to power law growth where
 $L_c$
grows with a power law having an exponent somewhere between
$L_c$
grows with a power law having an exponent somewhere between
 $1/5$
and
$1/5$
and
 $1/3$
, depending on the relative sizes of
$1/3$
, depending on the relative sizes of
 $\varGamma _1$
and
$\varGamma _1$
and
 $\varGamma _2$
.
$\varGamma _2$
.
We note here the difference between (D19) for the bending-dominated case as opposed to the membrane tension-dominated case, for which there is only one line tension between the two phases,
 $\varGamma =\int ^\infty _{-\infty } ( {\partial h}/{\partial g})^2\,{\rm d}g$
, with the growth law
$\varGamma =\int ^\infty _{-\infty } ( {\partial h}/{\partial g})^2\,{\rm d}g$
, with the growth law
 $ {{\rm d}L_c}/{{\rm d}t}\sim {\varGamma }/{L_c^2}$
. Both the terms in the bending-driven coarsening law are thus distinct from the membrane tension-driven coarsening law, because although
$ {{\rm d}L_c}/{{\rm d}t}\sim {\varGamma }/{L_c^2}$
. Both the terms in the bending-driven coarsening law are thus distinct from the membrane tension-driven coarsening law, because although
 $\varGamma _2$
is equivalent to
$\varGamma _2$
is equivalent to
 $\varGamma$
, it multiplies the curvature cubed in (D19) rather than the curvature as is the case for tension-driven coarsening (Bray Reference Bray1994).
$\varGamma$
, it multiplies the curvature cubed in (D19) rather than the curvature as is the case for tension-driven coarsening (Bray Reference Bray1994).


































































































 Open access
Open access