


Introduction
The cell is a crowded and complex environment where various physical forces operate within the genome and modify its organization and function. For example, crowding gives rise to depletion forces that tend to favor aggregates and condensates (Asakura and Oosawa, Reference Asakura and Oosawa1958; Marenduzzo et al., Reference Marenduzzo, Finan and Cook2006; Marenduzzo et al., Reference Marenduzzo, Finan and Cook2006; Renko, Reference Renko2010; Mardoum et al., Reference Mardoum, Gorczyca, Regan, Wu and Robertson-Anderson2018; Collette et al., Reference Collette, Dunlap and Finzi2023). Variations in crowding may, therefore, induce liquid-liquid phase separation (André and Spruijt, Reference André and Spruijt2020; Kohata and Miyoshi, Reference Kohata and Miyoshi2020; Shakya et al., Reference Shakya, Park, Rana and King2020; Gao et al., Reference Gao, Zhang, Chang, Zhang, Yang and Zhao2021; Collette et al., Reference Collette, Dunlap and Finzi2023) and confer a more gel- rather than fluid-like, or vice versa, nature to the genome (Odijk, Reference Odijk1998; Pelletier et al., Reference Pelletier, Halvorsen, Ha, Paparcone, Sandler, Woldringh, Wong and Jun2012; Ramisetty and Dias, Reference Ramisetty and Dias2015; Jorge et al., Reference Jorge, Nunes, Cova and Pais2016; Yang et al., Reference Yang, Männik, Retterer and Männik2020). Transitions from one phase to the other are expected to have significant regulatory implications for genomic transactions. It has been suggested that gel-like condensates may serve as factories or storage locations for DNA-processing enzymes, but also the genome may simply be more, or less, compacted, dynamic, and accessible to external molecules and compounds depending on the phase in which it is.
Other forces, constantly at play on the genome, are mechanical in nature due to the combination of tethering of DNA to membranes and the activities of processing enzymes acting as molecular machines that manipulate the double helix while consuming energy. These forces can pull and twist DNA, thereby changing its secondary structure and interaction with regulatory and remodeling proteins (Rouzina and Bloomfield, Reference Rouzina and Bloomfield2001; Li et al., Reference Li, Wijeratne, Qiu and Kiang2015; Svidlov et al., Reference Svidlov, Drobotenko, Basov, Gerasimenko, Malyshko, Elkina, Baryshev and Dzhimak2021; Collette et al., Reference Collette, Dunlap and Finzi2023). How entropic and mechanical forces, as well as their combined effect, change gene transcription is grossly understudied, despite the incredible insight that understanding the role of these forces would provide on transcription regulation.
Here, we report preliminary explorations of the effect of macromolecular crowding on: (i) LacI-mediated DNA looping topologies, (ii) Transcription rate, and (iii) Transcription termination given the following motivations.
Protein-mediated DNA looping is a ubiquitous mechanism of regulation, implicated in DNA replication, repair, recombination, and transcription. Since it brings distant segments of DNA close together, it is also an effective way to compact the genome (Amouyal, Reference Amouyal1991; Skoko et al., Reference Skoko, Yoo, Bai, Schnurr, Yan, McLeod, Marko and Johnson2006; Bouwman and de Laat, Reference Bouwman and de Laat2015). Protein-mediated DNA loops may be antiparallel (AP), or parallel (P). In the AP configuration, the DNA going into the loop has an opposite orientation from that exiting it and the DNA loop ‘extrudes’. In the P configuration, DNA has a solenoidal arrangement in space and the same orientation going in, or out, of the loop. When DNA wraps around proteins, such as in nucleosomes in eukaryotes or the 186 CI protein of the 186 bacteriophage, it clearly adopts a P configuration dictated by interactions with the proteins. In the case of loops secured only at the ends by proteins, the physiological significance of the P versus AP loop topologies is less clear, aside from the consideration that the solenoidal, P, is more compact than the extruded, AP, configuration.
In addition, crowding changes the compaction state of polymers. In particular, crowding affects the condensation state and phase of DNA. It may also change the viscosity of the environment altering the diffusion of reagents (Heinen et al., Reference Heinen, Zanini, Roosen-Runge, Fedunová, Zhang, Hennig, Seydel, Schweins, Sztucki, Antalík, Schreiber and Nägele2012; Skóra et al., Reference Skóra, Vaghefikia, Fitter and Kondrat2020; Aporvari et al., Reference Aporvari, Dang, Marfai, Coursey, McGorty and Robertson-Anderson2022). Thus, crowding may also affect the rate of transcript elongation. Finally, viscoelastic effects may affect the probability of termination versus either recycling (Harden et al., Reference Harden, Herlambang, Chamberlain, Lalanne, Wells, Li, Landick, Hochschild, Kondev and Gelles2020; Kang et al., Reference Kang, Ha, Uhm, Park, Lee, Hohng and Kang2020; Qian et al., Reference Qian, Wang, Artsimovitch, Dunlap and Finzi2024b) or readthrough (Ray-Soni et al., Reference Ray-Soni, Bellecourt and Landick2016; Vilborg and Steitz, Reference Vilborg and Steitz2017; Xie et al., Reference Xie, Libri and Porrua2023; Caldas et al., Reference Caldas, Luz, Baseggio, Andrade, Sobral and Grosso2024).
We tested these hypotheses using the tethered particle motion technique (Manzo and Finzi, Reference Manzo, Finzi and Nils2010; Manzo et al., Reference Manzo, Zurla, Dunlap and Finzi2012; Kumar et al., Reference Kumar, Manzo, Zurla, Ucuncuoglu, Finzi and Dunlap2014; Kovari et al., Reference Kovari, Yan, Finzi, Dunlap and Peterman2018a; Qian et al., Reference Qian, Collette, Finzi and Dunlap2024) and magnetic tweezers (Yan et al., Reference Yan, Ding, Leng, Dunlap and Finzi2018a; Yan et al., Reference Yan, Leng, Finzi and Dunlap2018b; Kovari et al., Reference Kovari, Dunlap, Weeks and Finzi2019; Piccolo et al., Reference Piccolo, Méndez, McCalla, Xu, Miller, Doan, Kovari, Dunlap and Finzi2021). These are single-molecule techniques particularly suitable to reveal conformational changes and the molecular details of dynamic processes such as RNA polymerase (RNAP) transcriptional elongation.
Materials and methods
DNA template preparation for TPM experiments
The DNA fragments used in these experiments were produced by polymerase chain reaction (PCR) as previously described (Qian et al., Reference Qian, Collette, Finzi and Dunlap2024). In brief, primers (Eurofins Genomics, Louisville, KY) designed with ‘A plasmid Editor’ (ApE) (Davis and Jorgensen, Reference Davis and Jorgensen2022) and labeled with biotin or digoxigenin, were used alongside deoxynucleotide triphosphates (New England BioLabs, NEB) and Taq/Q5 DNA polymerase (NEB). The resulting DNA fragments were attached at the digoxigenin end to the microscope flow chamber, and the biotin end to a bead.
Microchamber preparation for TPM experiments
Microchambers were prepared as previously reported (Qian et al., Reference Qian, Collette, Finzi and Dunlap2024). In brief, the lower microscope slide (Fisherbrand, Thermo Fisher Scientific, Waltham, MA) used to construct the microchamber supported a parafilm gasket that was cut to shape with a laser cutter (Universal Laser Systems, VLS 860, Middletown, CT). The central observation section was connected via narrow channels to inlet and outlet reservoirs just beyond the edges of the top coverslip. Once the chamber was assembled, it was heated to approximately 85°C on a hotplate and the top coverslip was pressed down with tweezers to seal the components together. The final volume was approximately 6 μL and provided a gradient of tether densities to optimize the utility of chambers. The sample preparation was conducted at room temperature (∼20°C) using λ buffer (10 mM Tris-HCl (pH 7.4), 200/100 mM KCl, 5% DMSO, 0.1 mM EDTA, 0.1 mg/mL α-casin, 0.2 mM DTT). Beads were polystyrene, streptavidin-coated, and 0.32 μm in diameter (Spherotech, Lake Forest, IL). Chambers were incubated with 10 μL anti-digoxigenin at a concentration of 5 μg/mL (Roche Life Science, Indianapolis, IN) in PBS at room temperature for 1 h. The anti-digoxigenin was then rinsed out with PBS and the chambers were then passivated with West-EZ Blocking Buffer (25 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% α-casin (w/v)) (GenDEPOT, Katy, TX) for 30 min. DNA tethers were then incubated in the chamber for 15 min to be anchored through a single digoxigenin to the anti-digoxigenin-coated coverslip. The other end of the DNA was anchored to a streptavidin-coated bead via a single biotin by adding 0.03 mg/mL beads solution in λ buffer for a 15 min incubation. Excess, untethered beads were then flushed out of the chamber with 120 μL of λ buffer. Beads were washed thrice in PBS and once in λ buffer before being resuspended in λ buffer prior to being introduced to the chamber. Lac repressor was added in a λ buffer solution containing different percentages (w/v) of crowder.
TPM procedure and analysis
Microspheres tethered to a glass substrate by individual DNA molecules undergo Brownian motion. The bead diffuses throughout a hemisphere, the size of which is dictated by the tether length. The scatter of positions of the tethered microsphere in the XY plane is recorded by single particle tracking to identify the anchor point and the extent of excursions around it. The excursion amplitude, ρ, value can be converted to the effective tether length using an appropriate calibration curve. The technique is reviewed in (Beausang et al., Reference Beausang, Zurla, Finzi, Sullivan and Nelson2007a, Reference Beausang, Zurla, Manzo, Dunlap, Finzi and Nelson2007b; Kovari et al., Reference Kovari, Yan, Finzi, Dunlap and Peterman2018a). All TPM measurements were performed as previously described (Yan et al., Reference Yan, Leng, Finzi and Dunlap2018b; Xu et al., Reference Xu, Yan, Artsimovitch, Dunlap and Finzi2022; Qian et al., Reference Qian, Collette, Finzi and Dunlap2024).
The XY position of each bead was recorded at 50 Hz interlaced with a custom Lab VIEW (National Instruments, Austin, TX) program. Vibrational or mechanical drift in the position of each bead was eliminated by subtracting the average location of reference beads that were adhered to the substrate within the same field of view. The effective length of each tether was then calculated as
 $ <\rho {>}_{8s}=\sqrt{{\left(x-<x{>}_{8s}\right)}^2+{\left(y-<y{>}_{8s}\right)}^2} $
, in which < x >8s and < y >8s are eight-second moving averages representing the coordinates of the anchor point of a bead. The anchor point is determined by finding these average x and y positions. Changes in the effective DNA tether length are represented as changes in the excursion length of the bead. Any beads with (x, y) position distributions with a ratio of the major to minor axes >1.07 were discarded to exclude beads tethered by multiple DNA molecules. Excursion data from the time records of beads, in the same experimental conditions, which passed the ‘symmetry test’ were retained for the analysis described below.
$ <\rho {>}_{8s}=\sqrt{{\left(x-<x{>}_{8s}\right)}^2+{\left(y-<y{>}_{8s}\right)}^2} $
, in which < x >8s and < y >8s are eight-second moving averages representing the coordinates of the anchor point of a bead. The anchor point is determined by finding these average x and y positions. Changes in the effective DNA tether length are represented as changes in the excursion length of the bead. Any beads with (x, y) position distributions with a ratio of the major to minor axes >1.07 were discarded to exclude beads tethered by multiple DNA molecules. Excursion data from the time records of beads, in the same experimental conditions, which passed the ‘symmetry test’ were retained for the analysis described below.
The excursion data was then fitted by a Bayesian step-filtering algorithm, which proved efficient and effective in processing step-like high-frequency signals, in this case, real-time signals with multiple fixed excursion levels corresponding to different looped and unlooped states. In brief, this method preceeds global filtering that finds an optimal time series
 $ {\rho}_{opt}(t) $
that eliminates the noise but retains the piecewise constant stepwise signals in the original time series
$ {\rho}_{opt}(t) $
that eliminates the noise but retains the piecewise constant stepwise signals in the original time series
 $ {\rho}_{ori}(t) $
. For this goal, we search for
$ {\rho}_{ori}(t) $
. For this goal, we search for
 $ {\rho}_{opt}(t) $
that minimizes the following cost function:
$ {\rho}_{opt}(t) $
that minimizes the following cost function:
 $ D\left({\rho}_{opt}|{\rho}_{ori}\right)=\frac{1}{2}\sum {\left({\rho}_{opt}(t)-{\rho}_{ori}(t)\right)}^2+\lambda \sum \left|{\rho}_{opt}\left(t+1\right)-{\rho}_{opt}(t)\right| $
, where λ is a parameter that determines the weights of the mean square error term and the total variation term. The details of minimizing the cost function have been described previously (Qian, Collette et al., Reference Qian, Collette, Finzi and Dunlap2024). After this process, the original excursion time series were smoothed as piecewise signals with alternating excursion levels. Finally, we assign the corresponding looped and unlooped states to each piece according to its excursion level and acquire the fraction of each state.
$ D\left({\rho}_{opt}|{\rho}_{ori}\right)=\frac{1}{2}\sum {\left({\rho}_{opt}(t)-{\rho}_{ori}(t)\right)}^2+\lambda \sum \left|{\rho}_{opt}\left(t+1\right)-{\rho}_{opt}(t)\right| $
, where λ is a parameter that determines the weights of the mean square error term and the total variation term. The details of minimizing the cost function have been described previously (Qian, Collette et al., Reference Qian, Collette, Finzi and Dunlap2024). After this process, the original excursion time series were smoothed as piecewise signals with alternating excursion levels. Finally, we assign the corresponding looped and unlooped states to each piece according to its excursion level and acquire the fraction of each state.
Proteins
LacI was provided by Kathleen Matthews (Rice University). HA-labeled E. coli RNA polymerase was provided by Karen Adelman (Harvard University).
DNA template preparation for MTs experiments
DNA tethers were prepared using plasmid pZV_NI_400 by performing PCR amplification with Q5 Hot start High-Fidelity master mix (New England Biolabs, Ipswich, MA). Single biotin-labeled forward primer, 5′-ATCGTTGGGAACCGGAG, and unlabeled reverse primer, 5′-AGCTTGTCTGTAAGCGGATG, were used to amplify 3 kb DNA fragments. The transcribed region contained the T7A1 promoter 741 bp from the lac operator, O1. The λt1 terminator was 612 bp downstream of O1 (Qian et al., Reference Qian, Collette, Finzi and Dunlap2024). The attachment point of the DNA to the bead surface was located at 1021 bp from the transcription termination site (Figure 1).
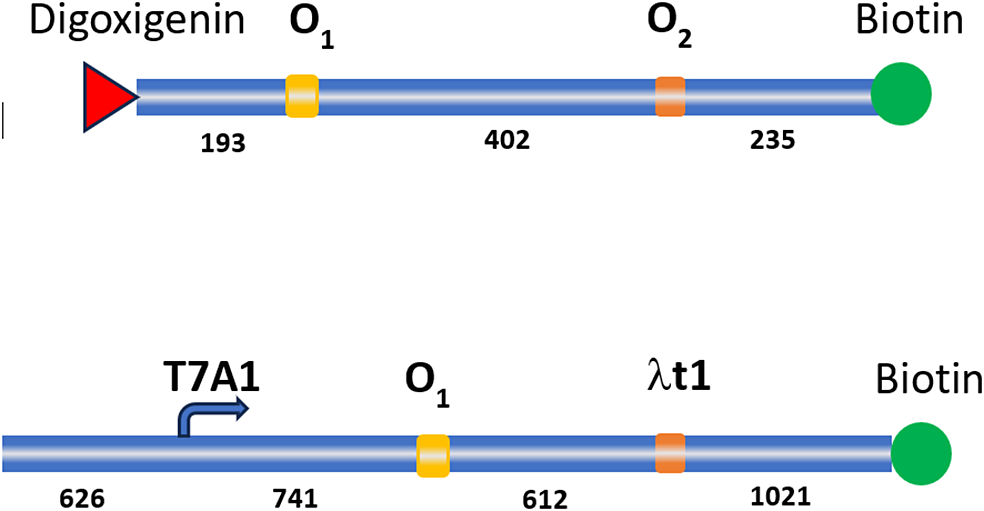
Figure 1. Schematic representation of DNA constructs used in TPM (top) and magnetic tweezers (bottom) measurements.
Microchamber preparation for MTs experiments
A laser-cut parafilm gasket was placed in the center of a clean, rectangular glass coverslip and covered with a square coverslip (Kovari et al., Reference Kovari, Yan, Finzi, Dunlap and Peterman2018a; Qian et al., Reference Qian, Collette, Finzi and Dunlap2024). The microchamber was heated for 15–20 s at 70°C on a hot plate to prevent leaks. Next, the anti-HA antibody was introduced to achieve a final concentration of 8 μg/mL into the microchamber and incubated for 30 min at room temperature. The microchamber was passivated with 15 μL blocking buffer (PBS with 1% casein, GeneTex, Irvine, CA) and incubated for 20 min at room temperature. Transcription elongation complexes (TECs) were stalled at position +21. They were prepared in an Eppendorf tube by mixing 3 nM DNA template, 30 nM HA-RNAP, 100 μM GpA dinucleotide (TriLink, San Diego, CA), 5 mM AUG in transcription buffer (20 mM Tris glutamate pH8, 50 mM potassium glutamate, 10 mM magnesium glutamate, 1 mM DTT, 0.2 mg/ml casein) and incubated for 10 min at 37°C. TECs were then diluted to 250 pM DNA:RNAP, added into the microchamber, and incubated for 10 min at room temperature. Next, 20 μL streptavidin-coated superparamagnetic beads (diluted 1:100 in Transcription Buffer; MyOneT1 Dyna beads, Life Technologies, Carlsbad, CA) were added into the microchamber and incubated for 5 min at room temperature. Finally, the microchamber was flushed with 20 μL transcription buffer to remove the excess streptavidin-coated superparamagnetic beads.
Procedure for magnetic tweezers experiments
Magnetic Tweezers were used to observe transcription and measure transcript elongation rates in real-time by monitoring the change in bead height as RNAP moved along the DNA template (Figure 2). Non-moving beads were selected as reference and moving beads for transcription assay (over 10 beads). Then, 50 μM NTPs (New England Biolabs, Ipswich, MA) were added into the microchamber without interrupting the recording which was stopped after 1 h for following data analysis. For crowding measurements, NTPs were added in a solution of polyethylene glycol (PEG). For percentages of PEG above 5%, the addition had to be done step by step with NTPs present only in the highest PEG concentration.
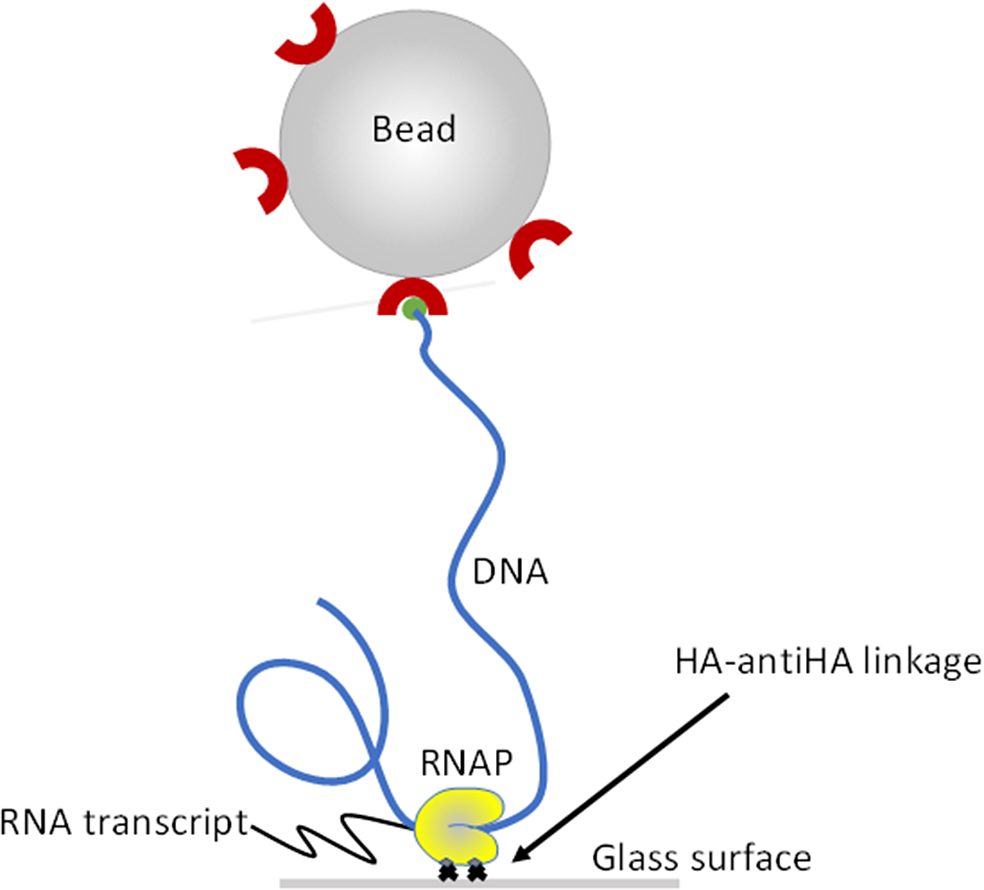
Figure 2. Schematic representation of the transcription assay set-up. Doubly HA-labeled RNA polymerase (RNAP; yellow) was attached to the microscope flow-chamber via a HA-anti-AH interaction. Single DNA molecules (blue) part of a DNA-RNAP ternary complex were labeled with a 1 mm-diameter, streptavidin-coated bead and served as templates for transcription assays.
Results
In vitro measurements are arguably the simplest and, sometimes, the only way to study molecular interactions that may be obscured in vivo. However, most in vitro measurements are made in saline solutions that are far from mimicking the native crowded environment of cells. The cell contains many molecular species, including membrane-bound and membrane-less organelles, macromolecules, and small molecules. Therefore, crowding is likely to exert entropic forces on molecules and macromolecules and affect intra-, as well as inter-, molecular interactions, including those relevant to transcription and its regulation. To begin addressing the gap between in vivo and in vitro measurements by characterizing the effect of macromolecular crowding on cellular processes, (i) DNA looping by the lac repressor (LacI) protein and (ii) transcription elongation by E. coli RNA polymerase (RNAP) were studied in the presence of a variety of polymeric, macromolecular crowders.
The tethered particle motion (TPM) technique (Kovari et al., Reference Kovari, Yan, Finzi, Dunlap and Peterman2018a; Qian et al., Reference Qian, Collette, Finzi and Dunlap2024) was used to monitor loop formation by the lac repressor protein (LacI) in a DNA template where two copies of the wild-type lac repressor binding site O1 were separated by ~400 base pairs (Figure 1). Dextran70 (Dx70, 70 kDa in molecular weight) at 2.5% and 5% weight fraction in solutions containing either 100 or 200 mM KCl, or Bovin Serum Albumin (BSA) at 5% and 10% fraction were used as crowders and introduced simultaneously to the lac repressor protein. PEG was not employed in these looping measurements because it caused the sticking of the bead to the surface as previously reported, possibly due to the collapse of the DNA tether (Lin et al., Reference Lin, Wuite and Dame2020). When LacI was introduced in the microchamber in a solution containing the crowder, looping was observed as a reversible shortening of the DNA tether. Both the parallel and antiparallel DNA looped conformations were observed (Yan et al., Reference Yan, Leng, Finzi and Dunlap2018b) and (Virnik et al., Reference Virnik, Lyubchenko, Karymov, Dahlgren, Tolstorukov, Semsey, Zhurkin and Adhya2003, Semsey et al., Reference Semsey, Tolstorukov, Virnik, Zhurkin and Adhya2004, Semsey et al., Reference Semsey, Virnik and Adhya2005, Lia Reference Lia, Semsey, Lewis, Adhya, Bensimon, Dunlap and Finzi2008, Olson et al., Reference Olson, Grosner, Czapla and Swigon2013) and the probability of each was calculated as described in Materials and Methods. Figure 3 shows that while the overall probability of looping was unchanged in different crowding conditions (unlooped percentages remained constant), anti-parallel looping decreased as parallel looping increased in crowded conditions.
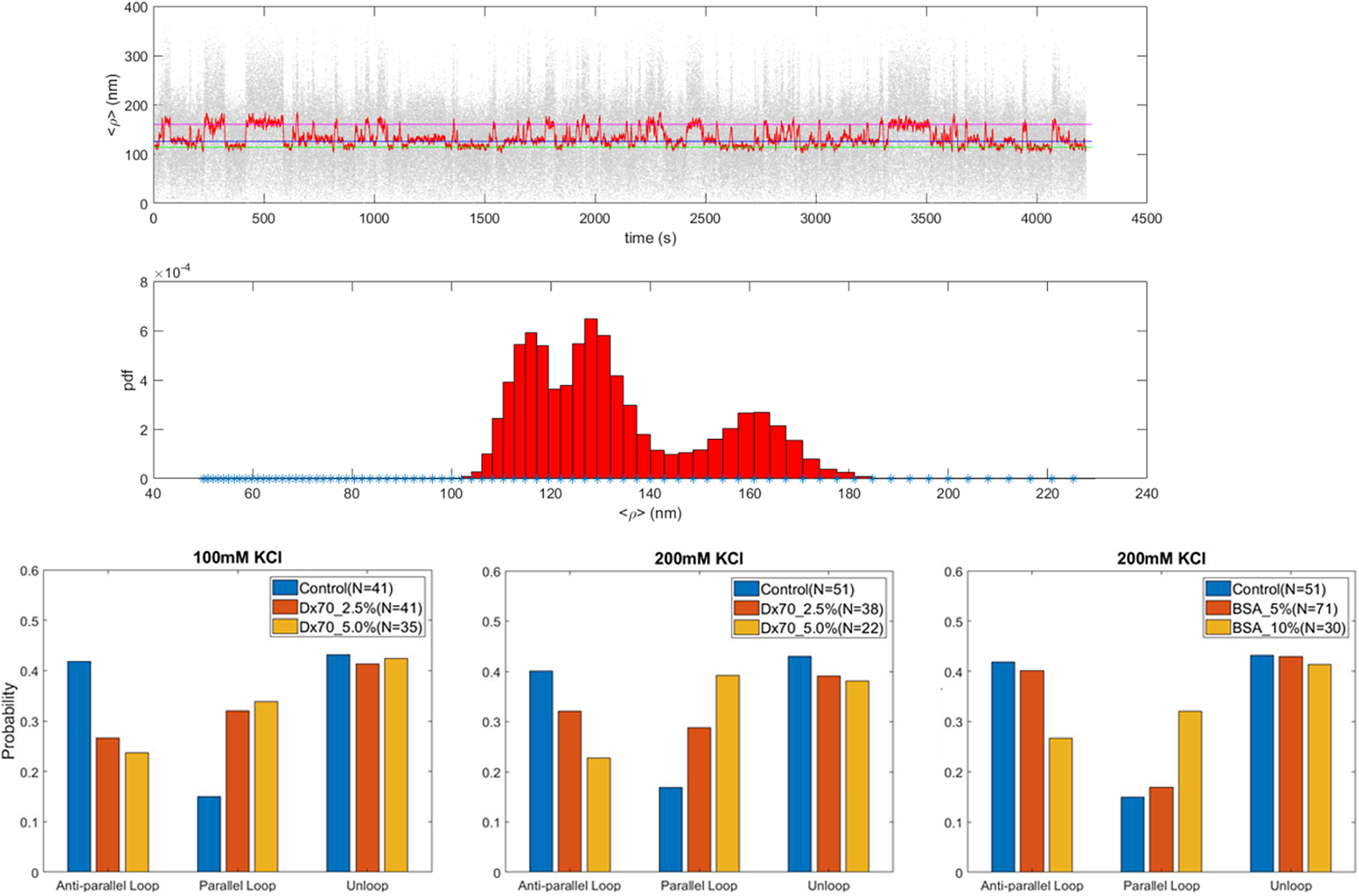
Figure 3. The antiparallel loop is disfavored in crowded conditions. (Top) Representative TPM trace and corresponding frequency distribution histogram showing the unlooped, parallel and antiparallel loop conformations (expected levels indicated by pink, blue and green lines each). (Bottom) Looping probabilities were plotted as described in Materials and Methods for the parallel and antiparallel DNA loops formed by the lac repressor in the presence of 100 mM (left) or 200 mM (center) KCl and different percentages of 70 kDa Dextran (Dx70), or 200 mM KCl and different percentages of Bovine Serum Albumin (BSA) (right). Different percentages are indicated by different colors with blue indicating the absence of crowder, red the lowest and yellow the highest percentage used.
The effect of macromolecular crowders on the rate of transcript elongation was probed using PEG of two molecular weights (2 and 8 kDa: PEG2000 and PEG8000) in the magnetic tweezers where a gentle tension was applied to the DNA (Smith et al., Reference Smith, Finzi and Bustamante1992; Strick et al., Reference Strick, Allemand, Croquette and Bensimon1998; Neuman and Nagy, Reference Neuman and Nagy2008; Seol and Neuman, Reference Seol and Neuman2011; Seol and Neuman, Reference Seol and Neuman2013; Kovari et al., Reference Kovari, Dunlap, Weeks and Finzi2019). This approach allowed monitoring transcription by RNAP in real time with better signal-to-noise levels avoiding DNA compaction. The top left panel in Figure 4 shows that increasing the concentration and molecular weight of PEG slowed down the rate of transcript elongation with the exception of 15% PEG2000.
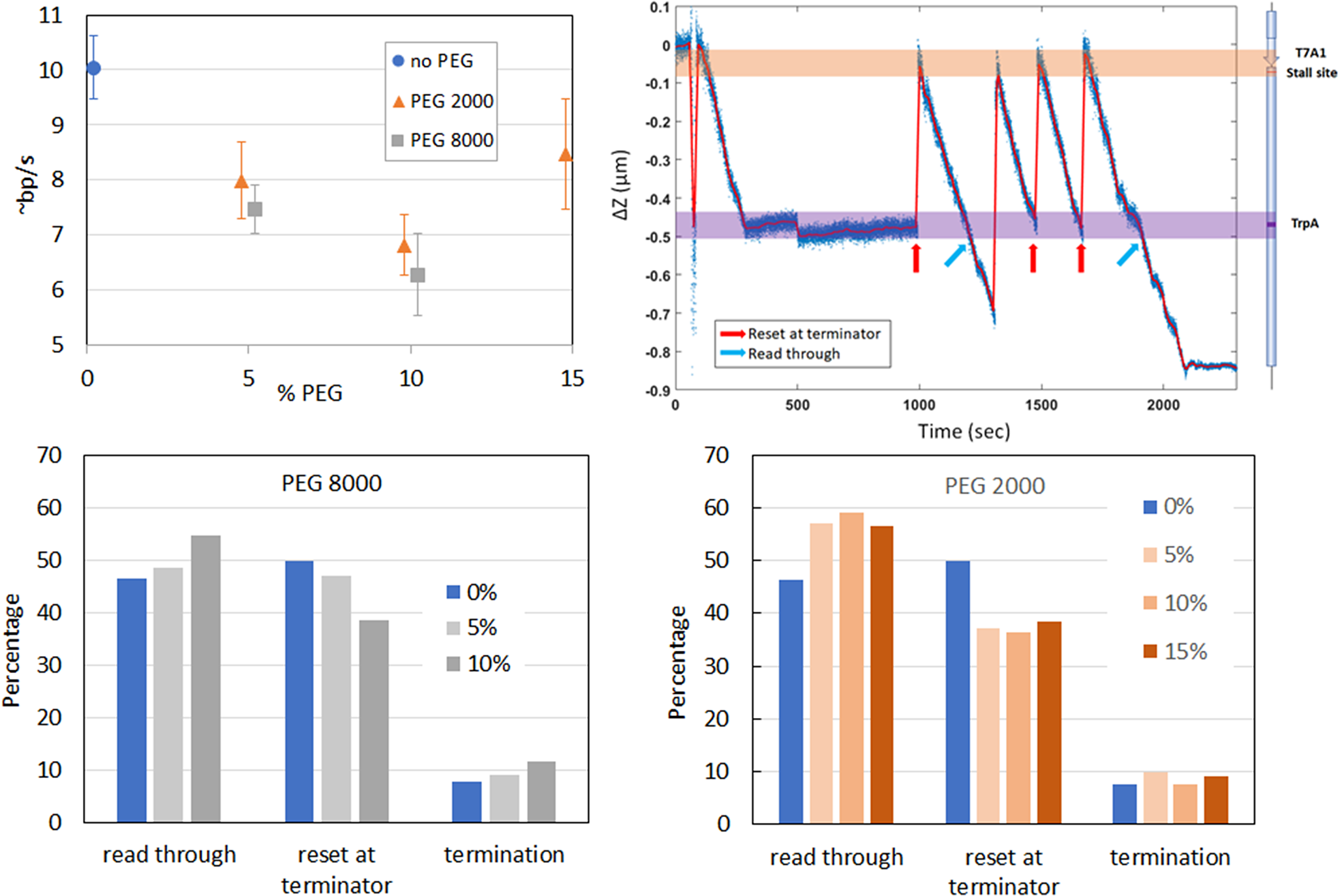
Figure 4. Effect of MMC on transcription. Rate of transcript elongation by E. coli RNAP in the presence of different percentages of PEG2000 or PEG8000 (Top left). Representative trace showing several rounds of transcription in the presence of PEG2000 (Top right). Relative probability for RNAP to (i) stop at the lt1 terminator (% stop at ter), performing another, or more, rounds of transcription once reached the terminator (% repeat trxn at ter), or (ii) read through (% read through) in various percentages of PEG2000 (Bottom left), or PEG8000 (Top right).
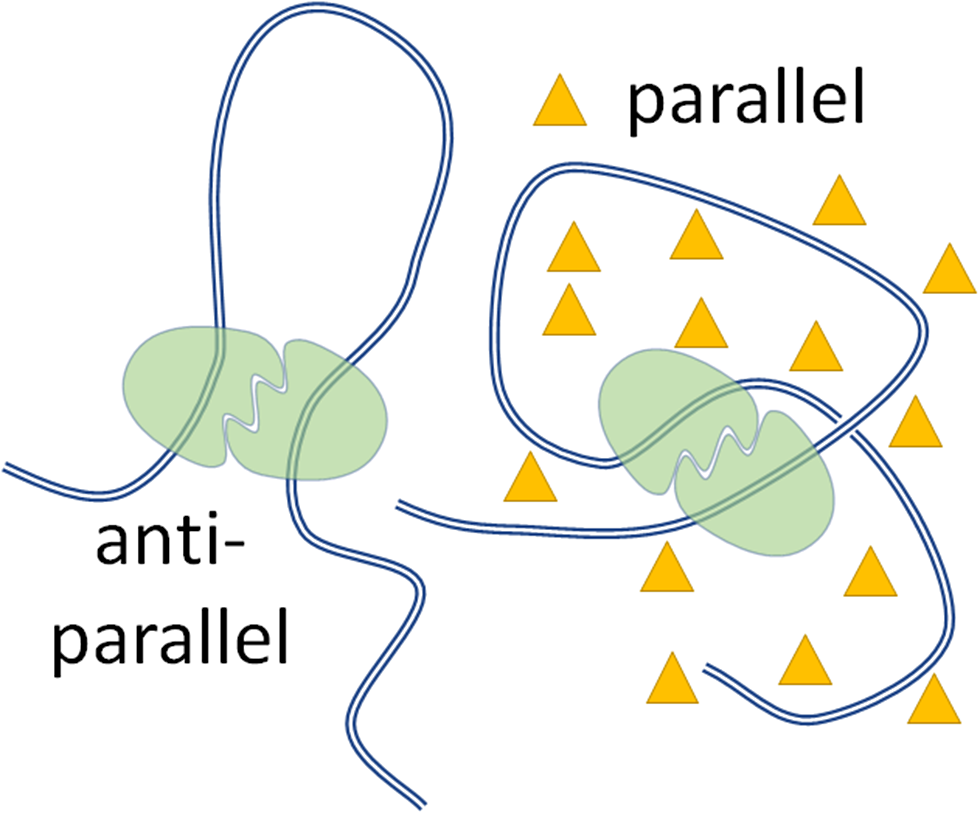
Figure 5. Schematic summary of the effect of MMC on protein-mediated DNA looping. Cartoon illustration showing the anti-parallel (left) and parallel (right) protein (green blobs)-induced DNA (blue lines) loops and the stabilization of the parallel loop by a crowder (yellow triangles).
The transcription traces also revealed that macromolecular crowding altered the probability of termination versus ‘run through’ (Figure 4 bottom), as well as that of transcription ‘recycling’ (Harden et al., Reference Harden, Herlambang, Chamberlain, Lalanne, Wells, Li, Landick, Hochschild, Kondev and Gelles2020; Kang et al., Reference Kang, Ha, Uhm, Park, Lee, Hohng and Kang2020; Qian et al., Reference Qian, Wang, Artsimovitch, Dunlap and Finzi2024b) (Figure 4 top right and bottom).
Discussion
The cell is an environment crowded with small and macro molecules where many processes take place simultaneously and, often, in competition. This limits the reach of in vivo experimentation to understand the structure, function, and physico-chemical properties of biomolecules in their native environment. On the other hand, the behavior of molecules and their biochemical interactions can be dissected with extreme precision in saline solutions in vitro. To bridge the informational gap between in vitro and in vivo measurements and enhance the relevance to the cellular scenario of the knowledge gathered from in vitro experiments, we monitored the dynamics of two processes, which are fundamental to the life of the cell: (i) protein-mediated DNA looping and (ii) DNA transcription, in crowded environments.
DNA looping by proteins which can bridge two, or multiple, sites is a broadly recognized, fundamental mechanism of regulation in almost all genomic transactions, such as DNA transcription, replication, repair, recombination, etc. A loop may form in either of two conformations, the parallel or the antiparallel, depending on the relative orientation of the DNA double helix going in and out of the loop closure point. The antiparallel conformation ‘consumes’ more DNA than the parallel conformation and, perhaps, exposes the loop more to the external environment. On the other hand, a parallel loop has a solenoidal geometry which may favor more compact packing with long helical fibers (Bancaud et al., Reference Bancaud, Silva, Barbi, Wagner, Allemand, Mozziconacci, Lavelle, Croquette, Victor, Prunell and Viovy2006). Our finding that crowding the solution with either of two different polymers, Dx70 and BSA at two different salt concentrations (for Dx70), favors one loop conformation versus the other (Figure 3 and Figure 5) suggests that modulation between the two conformations may be attained with local variations in crowding fraction alone, independently of salt concentration, or even crowder chemo-physical properties. Although these results are preliminary and more crowders and ionic strengths should be explored, these results indicate that the amount of crowding of the local environment may have important regulatory consequences, controlling accessibility versus compaction of DNA in the genome.
The effect of crowding on DNA transactions, such as transcription and its regulation could, in principle, be also important. Our measurements do not inform on possible effects on transcription initiation but do show changes in transcription rate and the possibility of effects on transcription recycling and the probability of run-off transcription. Higher molecular weight PEG showed stronger effects than lower molecular weights, indicating that crowder size plays a role. A crowding-induced decrease in the rate of elongation may allow for check-point and repair mechanisms to take place. The effect of crowding by PEG on transcription recycling and termination remains to be further investigated with other macromolecular crowders.
In summary, the measurements reported here show that local crowding variations could have significant regulatory effects on several DNA transactions and that the field is primed for investigation.
Open peer review
To view the open peer review materials for this article, please visit http://doi.org/10.1017/qrd.2025.8.
Acknowledgments
LacI and doubly HA-labeled RNAP were generous gifts from Kathleen Matthews, Rice University, and Karen Adelman, Harvard University, respectively. Plasmids for these experiments were created by Derrica McCalla.
Author contribution
Wenxuan Xu, Dylan Collette and Jin Qian Co-First authors. Dylan Collette performed the TPM LacI-induced DNA looping measurements, Jin Qian analyzed them and made Figure 3, Wenxuan Xu run the magnetic tweezers experiments in the presence of PEG, analyzed the data, and made the panels in Figure 4. David Dunlap designed the plasmids and led the project with Laura Finzi. Laura Finzi conceived and co-led the project. All participated in the writing of the manuscript.
Financial support
This study was supported by the National Institutes of Health (NIH) grants R01GM084070 and R35GM149296 to Laura Finzi.
Competing interests
The authors declare none.






 Open access
Open access
Comments
Dear Dr. Haunch,
Thank you for inviting me to submit a manuscript to be considered for publication in the “Single Molecule Challenges in the 21st Century” special issue of Quarterly Reviews of Biophysics: Discovery. My colleagues and I are, hereby, submitting our manuscript entitled “Insights on the effect of macromolecular crowding on transcription and its regulation” for consideration. In it, we describe experiments that probe the effect of macromolecular crowding on two fundamental cellular processes: DNA looping by proteins and transcription. Our findings highlight one of the current challenges in the single molecule field, that is bridging the gap between in vivo and in vitro experimentation by including crowders in the solution to mimic the cellular environment.
We believe that our work is relevant to your special issue and deserves publication in it.
Best regards,
Laura Finzi