


Introduction
The retinoic acid-induced 1 (RAI1) gene, located on chromosome 17p11.2, encodes a nuclear protein that plays a crucial role in maintaining normal neuropsychic functions (Ref. Reference Elsea and Williams1). Reviewing the research history of RAI1 reveals its intricate association with two rare neurodevelopmental syndromes – Smith–Magenis syndrome (SMS) (OMIM: 182290) and Potocki–Lupski syndrome (PTLS) (OMIM: 610883) – which share overlapping features while also exhibiting distinct, syndrome-specific phenotypes (Refs Reference Rinaldi, Villa, Sironi, Garavelli, Finelli and Bedeschi2, Reference Neira-Fresneda and Potocki3). It has long been recognised that RAI1 is situated within the crucial gene region linked to both SMS and PTLS (Refs Reference Potocki, Shaw, Stankiewicz and Lupski4, Reference Potocki, Bi, Treadwell-Deering, Carvalho, Eifert, Friedman, Glaze, Krull, Lee, Lewis, Mendoza-Londono, Robbins-Furman, Shaw, Shi, Weissenberger, Withers, Yatsenko, Zackai, Stankiewicz and Lupski5). Until 2003, Slager et al. (Ref. Reference Slager, Newton, Vlangos, Finucane and Elsea6) identified frameshift mutations in RAI1 among SMS patients, confirming the central role of RAI1 in this syndrome.
The clinical manifestations of SMS and PTLS in paediatric patients are complex, encompassing a broad spectrum of neurological, psychiatric and behavioural abnormalities (Refs Reference Potocki, Bi, Treadwell-Deering, Carvalho, Eifert, Friedman, Glaze, Krull, Lee, Lewis, Mendoza-Londono, Robbins-Furman, Shaw, Shi, Weissenberger, Withers, Yatsenko, Zackai, Stankiewicz and Lupski5, Reference Korteling, Musch, Zinkstok and Boot7). As the Human Genome Project progresses, RAI1 has been gradually recognised as being associated with various neuropsychiatric diseases. For instance, expansion of cytosine–adenine–guanine (CAG) repeats in RAI1 has been implicated in modifying spinocerebellar ataxia (SCA) (Ref. Reference Hayes, Turecki, Brisebois, Lopes-Cendes, Gaspar, Riess, Ranum, Pulst and Rouleau8). Large-scale genetic analyses in autism spectrum disorder (ASD) cohorts have also pinpointed RAI1 as a susceptibility gene for ASD (Ref. Reference Trost, Thiruvahindrapuram, Chan, Engchuan, Higginbotham, Howe, Loureiro, Reuter, Roshandel, Whitney, Zarrei, Bookman, Somerville, Shaath, Abdi, Aliyev, Patel, Nalpathamkalam, Pellecchia, Hamdan, Kaur, Wang, MacDonald, Wei, Sung, Lamoureux, Hoang, Selvanayagam, Deflaux, Geng, Ghaffari, Bates, Young, Ding, Shum, D’Abate, Bradley, Rutherford, Aguda, Apresto, Chen, Desai, Du, MLY, Pullenayegum, Samler, Wang, Ho, Paton, Pereira, Herbrick, Wintle, Fuerth, Noppornpitak, Ward, Magee, Al Baz, Kajendirarajah, Kapadia, Vlasblom, Valluri, Green, Seifer, Quirbach, Rennie, Kelley, Masjedi, Lord, Szego, Zawati, Lang, Strug, Marshall, Costain, Calli, Iaboni, Yusuf, Ambrozewicz, Gallagher, Amaral, Brian, Elsabbagh, Georgiades, Messinger, Ozonoff, Sebat, Sjaarda, Smith, Szatmari, Zwaigenbaum, Kushki, Frazier, JAS, Fakhro, Fernandez, MES, Weksberg, Fiume, RKC, Anagnostou, Sondheimer, Glazer, Hartley and Scherer9). Furthermore, postmortem analyses of individuals with schizophrenia, bipolar disorder or major depression have demonstrated significantly elevated RAI1 expression in brain tissues (Ref. Reference Haybaeck, Postruznik, Miller, Dulay, Llenos and Weis10). Recent foundational research has delved into the potential pathogenic mechanisms of RAI1, offering further insights into its role in neuropsychiatric diseases (Refs Reference Huang, Guenthner, Xu, Nguyen, Schwarz, Wilkinson, Gozani, Chang, Shamloo and Luo11, Reference Mullegama, Alaimo, Fountain, Burns, Balog, Chen and Elsea12, Reference Huang, Wang, Allen, Klope, Hu, Shamloo and Luo13, Reference Chang, Kowalczyk, Fogerson, Lee, Haque, Adams, Wang, DeNardo, Tessier-Lavigne, Huguenard, Luo and Huang14, Reference Turco, Giovenale, Sireno, Mazzoni, Cammareri, Marchioretti, Goracci, Di Veroli, Marchesan, D’Andrea, Falconieri, Torres, Bernardini, Magnifico, Paone, Rinaldo, Della Monica, D’Arrigo, Postorivo, Nardone, Zampino, Onesimo, Leoni, Caicci, Raimondo, Binda, Trobiani, De Jaco, Tata, Ferrari, Cutruzzolà, Mazzoccoli, Ziviani, Pennuto, Vescovi and Rosati15).
Given the critical role of RAI1 in nervous system function, we performed a systematic review summarising recent advances in both clinical and basic research on RAI1 in SMS and PTLS while providing an overview of research progress on its involvement in SCA, ASD, schizophrenia, bipolar disorder and major depression. This review aims to enhance our understanding of RAI1’s involvement across these conditions, with a particular emphasis on promoting the early recognition and diagnosis of SMS and PTLS.
Methods
Search strategy
This systematic review followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines (Ref. Reference Page, McKenzie, Bossuyt, Boutron, Hoffmann, Mulrow, Shamseer, Tetzlaff, Akl, Brennan, Chou, Glanville, Grimshaw, Hrobjartsson, Lalu, Li, Loder, Mayo-Wilson, McDonald, McGuinness, Stewart, Thomas, Tricco, Welch, Whiting and Moher16) (Supplementary Material 1). The final search was conducted in the PubMed and EMBASE databases on 1 October 2024. The search strategy used the terms ‘Retinoic acid-induced 1’ OR ‘RAI1’ in the ‘All Fields’ option to ensure comprehensive identification of relevant studies. Additionally, the reference lists of pertinent reviews and articles were manually searched to capture any potentially missed references.
The review protocol was registered in PROSPERO (CRD42023474165) and is available at https://www.crd.york.ac.uk/PROSPERO/#recordDetails.
Study selection
Articles were considered eligible if they met the following criteria: (1) original research conducted in English, (2) reporting on RAI1 in neuropsychiatric diseases, including SMS, PTLS, SCA, ASD, schizophrenia, bipolar disorder and major depression, (3) providing explicit details on genetic and phenotypic information for case reports and case series studies and (4) published before the last search date (1 October 2024). Two authors (T.Y. and D.P.) independently evaluated the titles, abstracts and full texts of the identified articles to determine eligibility. Any discrepancies were resolved through discussion or consultation with a third author if necessary.
Data extraction and quality assessment
The included studies were categorised into clinical and basic research based on the research theme. Clinical studies were defined as those involving patient cohorts, genetic analyses or phenotypic characterisations, whereas basic research included studies investigating molecular mechanisms, cellular functions and animal models of RAI1. The quality of all clinical studies was assessed using the Joanna Briggs Institute Critical Appraisal Checklist, where applicable (Ref. 17). Since SMS and PTLS patients share overlapping neurological and psychiatric symptoms, a standardised extraction form was used to collect detailed genotype and phenotype information for each case. This included the first author’s name, publication year, data source (country), disease, study design, sample size, gene sequencing method, variation pattern, deletion/duplication size (Mb), fragment location, mutation site, experimental validation of RAI1 transcription and expression changes, coexisting mutations, sex, age, clinical features and auxiliary examinations. Positive clinical phenotypes were recorded when explicitly stated in the original articles, whereas negative or unspecified phenotypes were treated as negative. Gene positions were standardised according to the human reference genome GRCh37 (hg19). For basic research studies, the focus was on investigating the effects of RAI1 deletion or duplication in animal models, along with the regulatory networks upstream and downstream of RAI1.
Statistics
Chi-square tests were performed to compare categorical clinical phenotype variables between SMS and PTLS patients. A two-sided p-value of <0.05 was considered statistically significant.
Results
Studies retrieved and characteristics
Database searches identified 569 records. After removing duplicates (n = 201) and screening titles and abstracts (n = 210), 158 full-text articles were assessed. Ultimately, 99 articles met the eligibility criteria and were included (Figure 1). These comprised 60 clinical studies (34 on SMS, 16 on PTLS, 5 on SCA, 4 on ASD and 1 on schizophrenia, bipolar disorder and major depression), and 39 basic research studies. Full details of quality assessment for the included clinical studies were available in Supplementary Material 2.
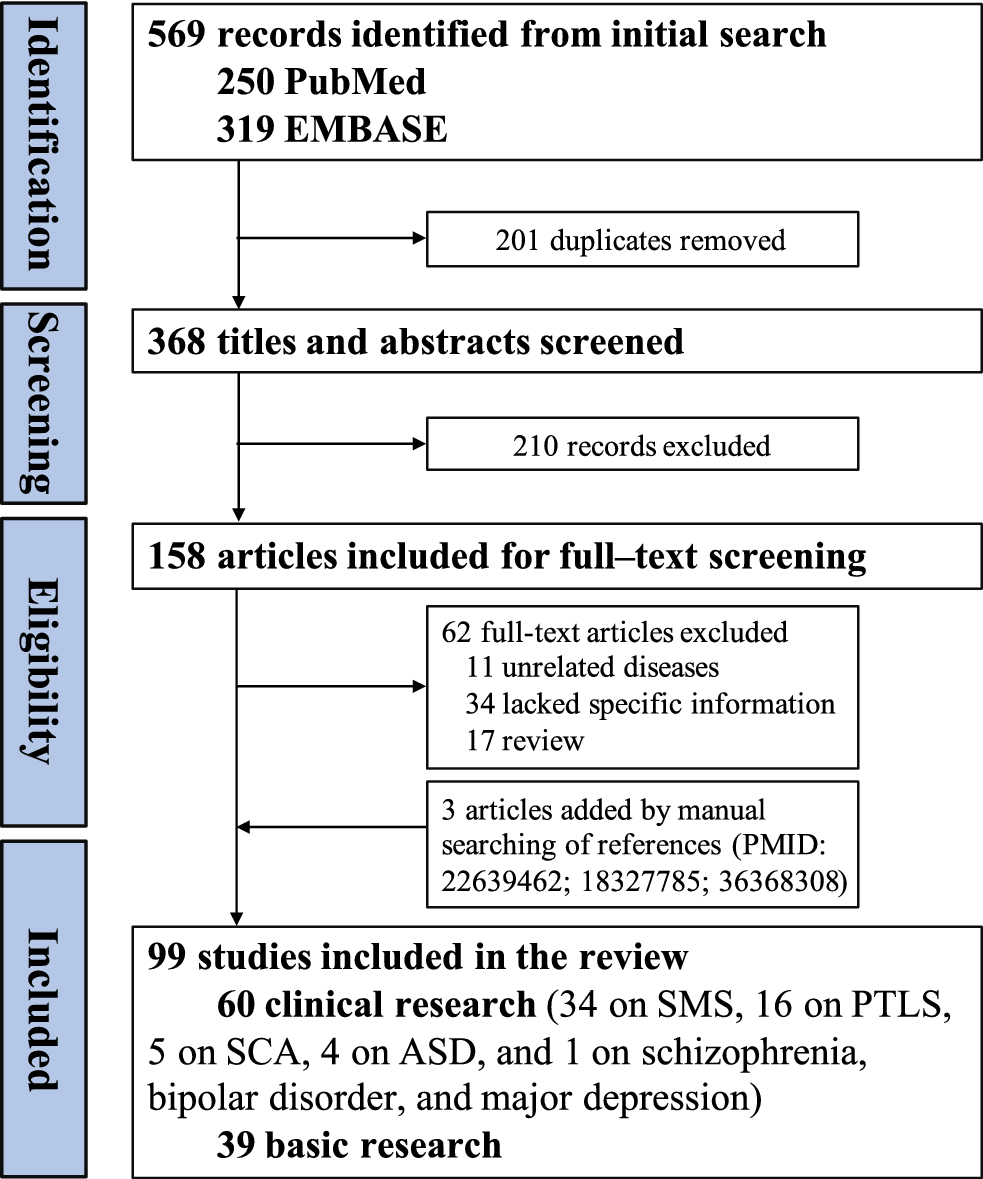
Figure 1. Flowchart of study selection.
ASD: autism spectrum disorder; PTLS: Potocki–Lupski syndrome; SCA: spinocerebellar ataxia; SMS: Smith–Magenis syndrome.
A total of 83 SMS patients and 64 PTLS patients were included in this review, with no overlapping cases. Detailed variant and phenotypic information is provided in Supplementary Material 3. To date, this represents the largest and most comprehensive summary of SMS and PTLS cases. Notably, there is a marked geographic imbalance in case distribution, with approximately 51% (42/83) of SMS cases and 67% (43/64) of PTLS cases reported from the United States (Refs Reference Potocki, Bi, Treadwell-Deering, Carvalho, Eifert, Friedman, Glaze, Krull, Lee, Lewis, Mendoza-Londono, Robbins-Furman, Shaw, Shi, Weissenberger, Withers, Yatsenko, Zackai, Stankiewicz and Lupski5, Reference Slager, Newton, Vlangos, Finucane and Elsea6, Reference Bi, Saifi, Shaw, Walz, Fonseca, Wilson, Potocki and Lupski18, Reference Myers and Challman19, Reference Girirajan, Elsas, Devriendt and Elsea20, Reference Vlangos, Wilson, Blancato, Smith and Elsea21, Reference Bi, Saifi, Girirajan, Shi, Szomju, Firth, Magenis, Potocki, Elsea and Lupski22, Reference Girirajan, Mendoza-Londono, Vlangos, Dupuis, Nowak, Bunyan, Hatchwell and Elsea23,Reference Nakamine, Ouchanov, Jiménez, Manghi, Esquivel, Monge, Fallas, Burton, Szomju, Elsea, Marshall, Scherer and McInnes24, Reference Zhang, Potocki, Sampson, Liu, Sanchez-Valle, Robbins-Furman, Navarro, Wheeler, Spence, Brasington, Withers and Lupski25, Reference Sanchez-Valle, Pierpont and Potocki26, Reference Sanford, Bermudez-Wagner, Jeng, Rauen and Slavotinek27, Reference Vilboux, Ciccone, Blancato, Cox, Deshpande, Introne, Gahl, Smith and Huizing28, Reference Goh, Perez, Canales, Ruiz, Agatep, Yoon, Chitayat, Dror, Shago, Goobie, Sgro, Walz and Mendoza-Londono29, Reference Adams, Yuan, Holyoak, Arajs, Hakimi, Markello, Wolfe, Vilboux, Burton, Fajardo, Grahame, Holloman, Sincan, Smith, Wells, Huang, Vega, Snyder, Golas, Tifft, Boerkoel, Hanson, Traynelis, Kerr and Gahl30, Reference Magoulas, Liu, Gelowani, Soler-Alfonso, Kivuva, Lupski and Potocki31, Reference Alaimo, Mullegama, Thomas and Elsea32, Reference Thaker, Esteves, Towne, Brownstein, James, Crowley, Hirschhorn, Elsea, Beggs, Picker and Agrawal33, Reference Yuan, Harel, Gu, Liu, Burglen, Chantot-Bastaraud, Gelowani, Beck, Carvalho, Cheung, Coe, Malan, Munnich, Magoulas, Potocki and Lupski34, Reference Yeetong, Vilboux, Ciccone, Boulier, Schnur, Gahl, Huizing, Laje and Smith35, Reference Yuan, Neira, Gu, Harel, Liu, Briceño, Elsea, Gómez, Potocki and Lupski36, Reference Abad, Cook, Cao, Jones, Rao, Dukes-Rimsky, Pauly, Skinner, Wang, Luo, Stevenson, Walz and Srivastava37, Reference Kuroda, Ritter, Mullegama and Izumi38, Reference Cuk, Unal, Jandric, Hayes, Walker, Abraamyan, Gornik and Ghazani39), and 24% (20/83) of SMS cases and 27% (17/64) of PTLS cases from Europe (Refs Reference Spadoni, Colapietro, Bozzola, Marseglia, Repossi, Danesino, Larizza and Maraschio40, Reference Schoumans, Staaf, Jönsson, Rantala, Zimmer, Borg, Nordenskjöld and Anderlid41, Reference Doco-Fenzy, Holder-Espinasse, Bieth, Magdelaine, Vincent, Khoury, Andrieux, Zhang, Lupski, Klink, Schneider, Goze-Martineau, Cuisset, Vallee, Manouvrier-Hanu, Gaillard and de Martinville42, Reference Capra, Biancheri, Morana, Striano, Novara, Ferrero, Boeri, Celle, Mancardi, Zuffardi, Parrini and Guerrini43, Reference Dubourg, Bonnet-Brilhault, Toutain, Mignot, Jacquette, Dieux, Gérard, Beaumont-Epinette, Julia, Isidor, Rossi, Odent, Bendavid, Barthélémy, Verloes and David44, Reference Acquaviva, Sana, Della Monica, Pinelli, Postorivo, Fontana, Falco, Nardone, Lonardo, Iascone and Scarano45, Reference Akkus, Kilic and Cubuk46, Reference Ciaccio, Pantaleoni, Milani, Alfei, Sciacca, Canafoglia, Erbetta and D’Arrigo47, Reference Grama, Sîrbe, Miclea, C clea, Huniadi, Bulata and Pop48, Reference Onesimo, Delogu, Blandino, Leoni, Rosati, Zollino and Zampino49, Reference Sironi, Bestetti, Masciadri, Tumiatti, Crippa, Pantaleoni, Russo, D’Arrigo, Milani, Larizza and Finelli50). In contrast, reports from Asia and Africa remain scarce. Thus, this work will improve awareness and facilitate the identification of RAI1-related diseases, particularly in underrepresented regions.
In basic research, mouse models for SMS and PTLS have successfully replicated many of the phenotypes observed in human patients (Refs Reference Huang, Guenthner, Xu, Nguyen, Schwarz, Wilkinson, Gozani, Chang, Shamloo and Luo11, Reference Walz, Caratini-Rivera, Bi, Fonseca, Mansouri, Lynch, Vogel, Noebels, Bradley and Lupski51, Reference Walz, Spencer, Kaasik, Lee, Lupski and Paylor52, Reference Bi, Ohyama, Nakamura, Yan, Visvanathan, Justice and Lupski53, Reference Girirajan, Patel, Slager, Tokarz, Bucan, Wiley and Elsea54, Reference Lacaria, Saha, Potocki, Bi, Yan, Girirajan, Burns, Elsea, Walz, Chan, Lupski and Gu55, Reference Cao, Molina, Abad, Carmona-Mora, Cárdenas Oyarzo, Young and Walz56). Preliminary studies have also explored potential therapeutic strategies for SMS and PTLS, identifying key therapeutic windows (Refs Reference Huang, Wang, Allen, Klope, Hu, Shamloo and Luo13, Reference Alaimo, Hahn, Mullegama and Elsea57, Reference Javed, Lee, Xu and Huang58, Reference Chang, Lee, Javed, Haque, Chang, Lin, Oram and Huang59) Notably, animal studies have provided valuable insights into the mechanisms underlying the complex phenotypes of these disorders, including abnormal body weight, sleep disturbances, circadian rhythm disruptions and seizures (Refs Reference Mullegama, Alaimo, Fountain, Burns, Balog, Chen and Elsea12, Reference Chang, Kowalczyk, Fogerson, Lee, Haque, Adams, Wang, DeNardo, Tessier-Lavigne, Huguenard, Luo and Huang14, Reference Lacaria, Saha, Potocki, Bi, Yan, Girirajan, Burns, Elsea, Walz, Chan, Lupski and Gu55, Reference Alaimo, Hahn, Mullegama and Elsea57, Reference Javed, Lee, Xu and Huang58, Reference Burns, Schmidt, Williams, Kim, Girirajan and Elsea60, Reference Williams, Zies, Mullegama, Grotewiel and Elsea61, Reference Lacaria, Gu and Lupski62, Reference Diessler, Kostic, Arsenijevic, Kawasaki and Franken63). The following section offers a comprehensive review and synthesis of these findings.
Clinical research on RAI1 in neuropsychiatric diseases
RAI1 in Smith–Magenis syndrome and Potocki–Lupski syndrome
SMS and PTLS are rare neurodevelopmental disorders that present unique challenges in paediatric care. Both conditions are caused by alterations in the dosage of the RAI1 gene (Refs Reference Rinaldi, Villa, Sironi, Garavelli, Finelli and Bedeschi2, Reference Neira-Fresneda and Potocki3). SMS results from RAI1 haploinsufficiency, typically due to 17p11.2 deletions (most commonly ~3.7 Mb) or pathogenic mutations, whereas PTLS is caused by RAI1 overexpression resulting from 17p11.2 microduplications (0.25–11.2 Mb) (Supplementary Material 3). To date, 43 pathogenic RAI1 point mutations have been identified in SMS patients (Supplementary Material 3). Among these, 51.2% are frameshift mutations, 23.3% are missense variants and 23.3% are nonsense mutations (Figure 2).
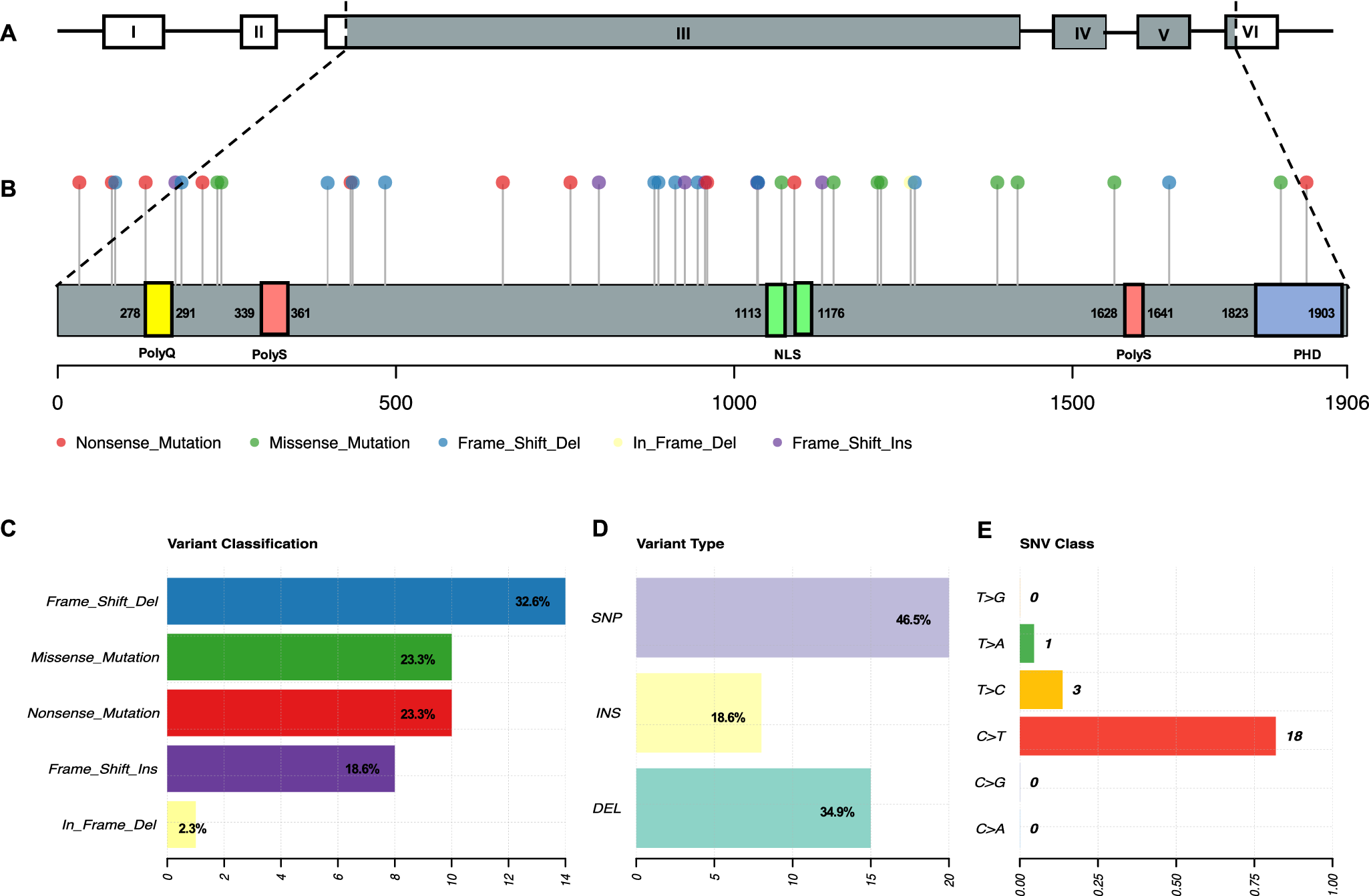
Figure 2. RAI1 point mutations in Smith–Magenis syndrome. A. RAI1 gene structure. B. RAI1 protein structure with the distribution of point mutations. C–E. Classification of RAI1 point mutations.
While SMS and PTLS share overlapping clinical features – such as craniofacial abnormalities, developmental delays, and behavioural challenges – distinct phenotypic patterns are evident (Refs Reference Rinaldi, Villa, Sironi, Garavelli, Finelli and Bedeschi2, Reference Neira-Fresneda and Potocki3). Based on the types of variations involved in the RAI1 gene, we summarised the clinical manifestations of SMS and PTLS patients in Table 1. Overall, SMS is often characterised by brachycephaly, midface hypoplasia, scoliosis, cognitive impairment, sleep disturbances, seizures, self-injurious behaviour and obesity. In contrast, PTLS is associated with micrognathia, slanting palpebral fissures, malformed ears, renal anomalies, underweight and muscle weakness.
Table 1. Phenotypic features in patients with SMS and PTLS
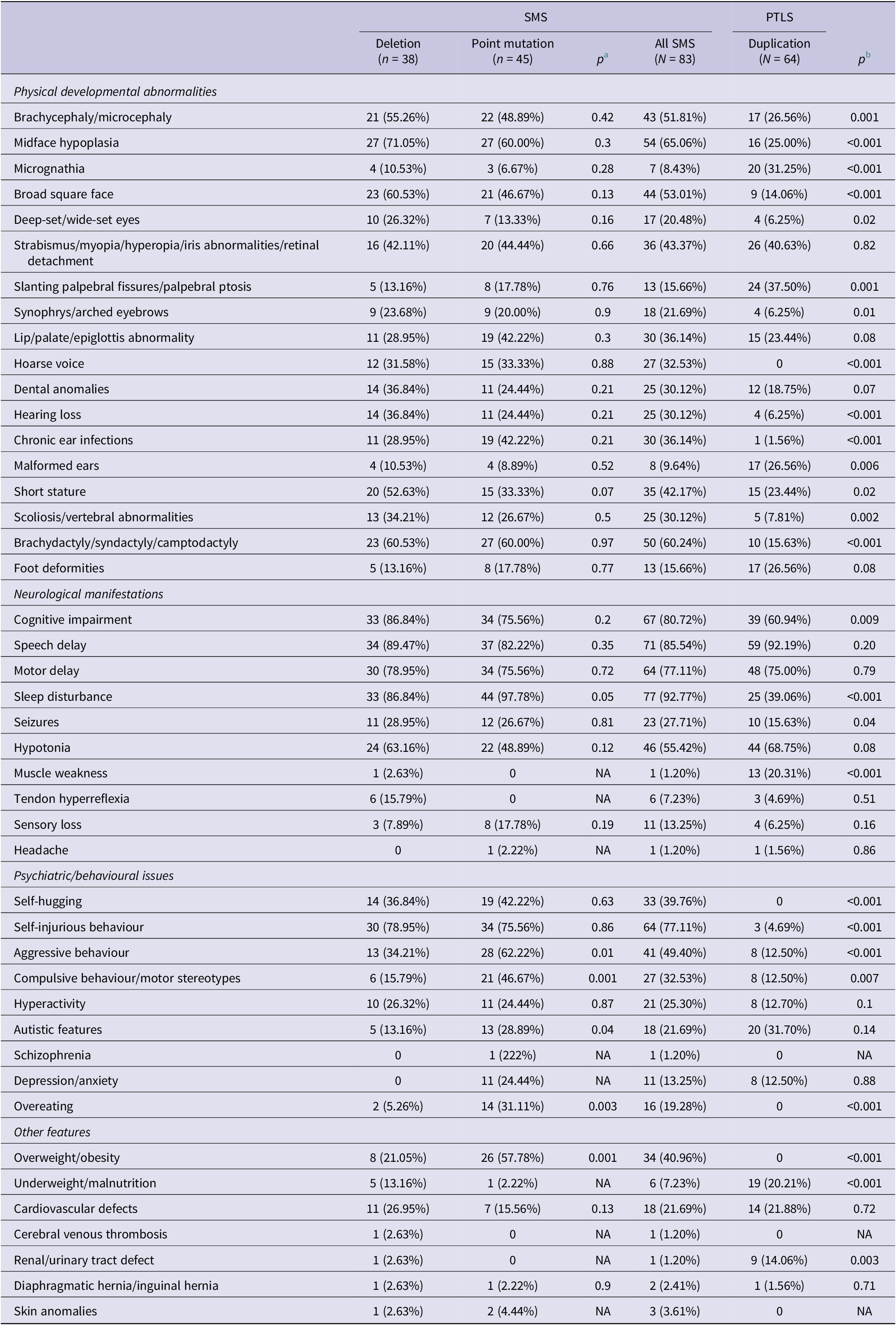
NA: not applicable; PTLS: Potocki–Lupski syndrome; SMS: Smith–Magenis syndrome.
a Comparison of phenotype between SMS patients with 17p11.2 deletion and RAl1 point mutation.
b Comparison of phenotype between all SMS patients and PTLS patients.
For paediatricians, recognising these differences is crucial for accurate diagnosis and tailored management. Of particular note, some phenotypes are syndrome-specific: overweight/obesity is observed only in SMS, whereas underweight/malnutrition and muscle weakness are primarily reported in PTLS. These findings emphasise the nuanced effects of RAI1 gene dosage, which exhibit overlapping but distinct impacts on neurodevelopment. Understanding these variations can guide early diagnosis, intervention and long-term care, especially in resource-limited settings where SMS and PTLS may be underdiagnosed.
In patients with SMS, physical development and neurological features are largely comparable between individuals carrying 17p11.2 deletions and those with RAI1 point mutations. However, patients with RAI1 mutations exhibit a higher prevalence of sleep disturbances, aggressive behaviour, compulsive behaviour/motor stereotypes, autistic traits, overeating and overweight/obesity compared with those with 17p11.2 deletions (Table 1). Notably, larger deletions spanning approximately 5–10 Mb may encompass RAI1-adjacent genes, such as PMP22, leading to PMP22-RAI1 deletions. Compared with the typical 3.7 Mb deletions seen in SMS, individuals with larger deletions are more likely to exhibit clinical signs of peripheral neuropathy and/or objective electrophysiological abnormalities (Supplementary Material 3), along with an earlier onset and increased severity of peripheral neuropathy (Ref. Reference Yuan, Neira, Gu, Harel, Liu, Briceño, Elsea, Gómez, Potocki and Lupski36). These findings suggest that variability in 17p11.2 deletion sizes affects not only RAI1 but also neighbouring genes, potentially contributing to distinct clinical phenotypes.
Most case studies have provided some clues regarding the variant patterns, RAI1 expression levels and their relationship to the diagnosis of SMS and PTLS. Typically, RAI1 deletions and nonsense mutations reduce gene expression and are linked to SMS, while duplications increase expression and lead to PTLS. However, exceptions, particularly involving missense mutations or regulatory region variants, challenge this framework. For instance, individuals carrying missense mutations in RAI1 (e.g., p.Y236F, p.T1061T, p.Q1389R and p.R1217Q) exhibit SMS features, yet Western blotting or quantitative PCR often shows unchanged RAI1 expression levels (Refs Reference Vilboux, Ciccone, Blancato, Cox, Deshpande, Introne, Gahl, Smith and Huizing28, Reference Vieira, Rodriguez, Carmona-Mora, Cao, Gamba, Carvalho, de Rezende Duarte, Santos, de Souza, DuPont, Walz, Moretti-Ferreira and Srivastava64). Similarly, a case with the p.R1147Q de novo mutation demonstrated a normal protein level and localisation but impaired transcriptional activation of brain-derived neurotrophic factor (BDNF), suggesting abnormal regulatory activity as a potential mechanism (Ref. Reference Abad, Cook, Cao, Jones, Rao, Dukes-Rimsky, Pauly, Skinner, Wang, Luo, Stevenson, Walz and Srivastava37).
PTLS cases also reveal complexity. One patient with no changes in RAI1 copy number carried a maternally inherited upstream deletion that disrupted regulatory elements, leading to elevated RAI1 mRNA levels (Ref. Reference Alaimo, Mullegama, Thomas and Elsea32). Conversely, an SMS case with a deletion involving exons 5 and 6 paradoxically resulted in increased RAI1 transcript levels due to loss of the canonical stop codon, replaced by a downstream alternative (Ref. Reference Sironi, Bestetti, Masciadri, Tumiatti, Crippa, Pantaleoni, Russo, D’Arrigo, Milani, Larizza and Finelli50). These unique cases underscore the nuanced relationship between RAI1 variant types, expression levels and functional activity. They also highlight diagnostic challenges in distinguishing SMS from PTLS and suggest that clinical evaluation should consider both variant patterns and functional consequences of RAI1 dysregulation.
RAI1 in spinocerebellar ataxia
SCA comprises a group of inherited disorders characterised by progressive degeneration of the cerebellum, brainstem and spinal cord (Ref. Reference Klockgether, Mariotti and Paulson65). Among its subtypes, SCA1 and SCA2 are caused by CAG repeat expansions in ATXN1 and ATXN2, where repeat length correlates with earlier disease onset (Refs Reference Klockgether, Mariotti and Paulson65, Reference Tezenas du Montcel, Durr, Bauer, Figueroa, Ichikawa, Brussino, Forlani, Rakowicz, Schols, Mariotti, van de Warrenburg, Orsi, Giunti, Filla, Szymanski, Klockgether, Berciano, Pandolfo, Boesch, Melegh, Timmann, Mandich, Camuzat, Goto, Ashizawa, Cazeneuve, Tsuji, Pulst, Brusco, Riess, Brice and Stevanin66). RAI1 contains a polymorphic CAG repeat tract and has been implicated in polyglutamine disorders (Ref. Reference Butland, Devon, Huang, Mead, Meynert, Neal, Lee, Wilkinson, Yang, Yuen, Hayden, Holt, Leavitt and Ouellette67). Studies suggest a potential role for RAI1 in modifying the onset of SCA. In a study of 46 SCA2 patients, the repeat size of RAI1 was found to explain an additional 4.1% of the variation in age of onset after accounting for the impact of ATXN2 expansions (Ref. Reference Hayes, Turecki, Brisebois, Lopes-Cendes, Gaspar, Riess, Ranum, Pulst and Rouleau8). Similarly, research on 23 independent families with SCA2 in India supported this finding, suggesting that RAI1 CAG repeat variation contributed to approximately 13% of the unexplained variation in age of onset for SCA2 (Ref. Reference Chattopadhyay, Ghosh, Gangopadhyay, Das, Roy, Sinha, Jha, Mukherjee, Chakraborty, Singhal, Bhattacharya and Bhattacharyya68). However, these results were not validated in a South American SCA2 cohort, where no association was found between RAI1 repeat size and disease onset or clinical phenotype (Ref. Reference Pereira, Monte, Locks-Coelho, Silva, Barsottini, Pedroso, Cornejo-Olivas, Mazzetti, Godeiro, Vargas, Lima, van der Linden, Toralles, Medeiros, Ribeiro, Braga-Neto, Salarini, Castilhos, Saraiva-Pereira and Jardim69). In SCA1, a study involving 152 patients in China found no significant modifying effect of RAI1 CAG repeats on disease onset (Ref. Reference Wang, Chen, Peng, Cao, Li, Wang, Yang, Peng, Shi, Zhou, Li, Feng, Wu, Qiu, Xia, Tang and Jiang70). While these studies provide preliminary evidence for a potential modifying role of RAI1, the overall link between RAI1 and SCA remains inconclusive and requires further investigation with larger and more diverse cohorts.
RAI1 in autism spectrum disorder, schizophrenia, bipolar disorder and major depression
Genetic studies from the Netherlands and France have identified RAI1 as a potential susceptibility gene for ASD (Refs Reference van der Zwaag, Franke, Poot, Hochstenbach, Spierenburg, Vorstman, van Daalen, de Jonge, Verbeek, Brilstra, van ’t Slot, Ophoff, van Es, Blauw, Veldink, Buizer-Voskamp, Beemer, van den Berg, Wijmenga, van Amstel, van Engeland, Burbach and Staal71, Reference Redin, Gérard, Lauer, Herenger, Muller, Quartier, Masurel-Paulet, Willems, Lesca, El-Chehadeh, Le Gras, Vicaire, Philipps, Dumas, Geoffroy, Feger, Haumesser, Alembik, Barth, Bonneau, Colin, Dollfus, Doray, Delrue, Drouin-Garraud, Flori, Fradin, Francannet, Goldenberg, Lumbroso, Mathieu-Dramard, Martin-Coignard, Lacombe, Morin, Polge, Sukno, Thauvin-Robinet, Thevenon, Doco-Fenzy, Genevieve, Sarda, Edery, Isidor, Jost, Olivier-Faivre, Mandel and Piton72). Network analyses of gene interactions have further revealed connections between RAI1 and multiple ASD-related proteins, suggesting their involvement in shared molecular pathways (Ref. Reference Mullegama, Alaimo, Chen and Elsea73). In the most recent and extensive global whole-genome sequencing study, which included 5,100 individuals with ASD and 6,212 non-ASD parents and siblings, identified RAI1 as significantly associated with ASD (Ref. Reference Trost, Thiruvahindrapuram, Chan, Engchuan, Higginbotham, Howe, Loureiro, Reuter, Roshandel, Whitney, Zarrei, Bookman, Somerville, Shaath, Abdi, Aliyev, Patel, Nalpathamkalam, Pellecchia, Hamdan, Kaur, Wang, MacDonald, Wei, Sung, Lamoureux, Hoang, Selvanayagam, Deflaux, Geng, Ghaffari, Bates, Young, Ding, Shum, D’Abate, Bradley, Rutherford, Aguda, Apresto, Chen, Desai, Du, MLY, Pullenayegum, Samler, Wang, Ho, Paton, Pereira, Herbrick, Wintle, Fuerth, Noppornpitak, Ward, Magee, Al Baz, Kajendirarajah, Kapadia, Vlasblom, Valluri, Green, Seifer, Quirbach, Rennie, Kelley, Masjedi, Lord, Szego, Zawati, Lang, Strug, Marshall, Costain, Calli, Iaboni, Yusuf, Ambrozewicz, Gallagher, Amaral, Brian, Elsabbagh, Georgiades, Messinger, Ozonoff, Sebat, Sjaarda, Smith, Szatmari, Zwaigenbaum, Kushki, Frazier, JAS, Fakhro, Fernandez, MES, Weksberg, Fiume, RKC, Anagnostou, Sondheimer, Glazer, Hartley and Scherer9). Evidence for RAI1’s involvement in schizophrenia, bipolar disorder and major depression is relatively limited. A post-mortem study reported significantly increased RAI1 expression in the dorsolateral prefrontal cortex of patients with these psychiatric disorders (Ref. Reference Haybaeck, Postruznik, Miller, Dulay, Llenos and Weis10). However, a genome-wide association study found no significant association between RAI1 and schizophrenia risk (Ref. Reference Singh, Poterba, Curtis, Akil, Al Eissa, Barchas, Bass, Bigdeli, Breen, Bromet, Buckley, Bunney, Bybjerg-Grauholm, Byerley, Chapman, Chen, Churchhouse, Craddock, Cusick, DeLisi, Dodge, Escamilla, Eskelinen, Fanous, Faraone, Fiorentino, Francioli, Gabriel, Gage, Gagliano Taliun, Ganna, Genovese, Glahn, Grove, Hall, Hamalainen, Heyne, Holi, Hougaard, Howrigan, Huang, Hwu, Kahn, Kang, Karczewski, Kirov, Knowles, Lee, Lehrer, Lescai, Malaspina, Marder, SA, AM, Medeiros, Milani, Morley, Morris, Mortensen, Myers, Nordentoft, O’Brien, Olivares, Ongur, Ouwehand, Palmer, Paunio, Quested, Rapaport, Rees, Rollins, Satterstrom, Schatzberg, Scolnick, Scott, Sharp, Sklar, Smoller, Sobell, Solomonson, Stahl, Stevens, Suvisaari, Tiao, Watson, Watts, Blackwood, Borglum, Cohen, Corvin, Esko, Freimer, Glatt, Hultman, McQuillin, Palotie, Pato, Pato, Pulver, St Clair, Tsuang, Vawter, Walters, Werge, Ophoff, Sullivan, Owen, Boehnke, O’Donovan, Neale and Daly74). These findings suggest that RAI1 is crucial for mental health, not only during early development but potentially throughout the lifespan, warranting further research into its broader neuropsychiatric implications.
Basic research on RAI1 in Smith–Magenis syndrome and Potocki–Lupski syndrome
Animal model studies have been instrumental in elucidating the essential role of RAI1 in mammalian development and homeostasis, shedding light on its involvement in SMS and PTLS. Walz et al. (Ref. Reference Walz, Caratini-Rivera, Bi, Fonseca, Mansouri, Lynch, Vogel, Noebels, Bradley and Lupski51) engineered mouse models carrying either a deletion (Df(11)17) or duplication (Dp(11)17) in a chromosomal region syntenic to human 17p11.2, commonly affected in SMS and PTLS. Df(11)17/+ mice exhibited craniofacial abnormalities, seizures, significant obesity and male-specific reduced fertility, whereas Dp(11)17/+ mice were underweight and did not experience seizures (Ref. Reference Walz, Caratini-Rivera, Bi, Fonseca, Mansouri, Lynch, Vogel, Noebels, Bradley and Lupski51). No apparent histological defects were observed in the hearts, spleens or kidneys of either model (Ref. Reference Walz, Caratini-Rivera, Bi, Fonseca, Mansouri, Lynch, Vogel, Noebels, Bradley and Lupski51), highlighting the specific phenotypic consequences of these alterations. To minimise confounding effects from neighbouring genes in the 17p11.2 region, researchers developed transgenic mice with altered Rai1 expression. The findings emphasised Rai1 as a dosage-sensitive gene critical for central nervous system function. Homozygous Rai1 −/− mice exhibited severe phenotypes, with most dying during early development (Refs Reference Bi, Ohyama, Nakamura, Yan, Visvanathan, Justice and Lupski53, Reference Bi, Yan, Shi, Yuva-Paylor, Antalffy, Goldman, Yoo, Noebels, Armstrong, Paylor and Lupski75). Rare survivors demonstrated growth retardation, craniofacial malformations, seizures, motor impairment and cognitive deficits (Refs Reference Bi, Ohyama, Nakamura, Yan, Visvanathan, Justice and Lupski53, Reference Bi, Yan, Shi, Yuva-Paylor, Antalffy, Goldman, Yoo, Noebels, Armstrong, Paylor and Lupski75). In contrast, heterozygous Rai1 +/− mice exhibited SMS-like traits, including craniofacial anomalies, obesity, seizures, learning difficulties and social behaviour abnormalities (Refs Reference Bi, Ohyama, Nakamura, Yan, Visvanathan, Justice and Lupski53, Reference Bi, Yan, Shi, Yuva-Paylor, Antalffy, Goldman, Yoo, Noebels, Armstrong, Paylor and Lupski75, Reference Rao, Abad, Perez, Srivastava, Young and Walz76).
Girirajan et al. (Ref. Reference Girirajan, Patel, Slager, Tokarz, Bucan, Wiley and Elsea54) graded overexpression of Rai1, finding that animals with 1.5- to 2-fold Rai1 overexpression showed PTLS-like features, such as growth retardation, reduced body weight, increased anxiety-like behaviours and heightened locomotor activity. Additionally, in Rai1 conditionally transgenic mice with Rai1 specifically overexpressed in the hippocampus, cerebral cortex, striatum, amygdala and olfactory bulb, significant weight loss was also observed (Ref. Reference Cao, Molina, Abad, Carmona-Mora, Cárdenas Oyarzo, Young and Walz56).
Besides these phenotypic studies of animal models, the mechanisms underlying Rai1 regulation of weight abnormalities, disrupted sleep rhythms and epileptic seizures have been investigated. Such studies provide foundational insights for understanding and potentially managing SMS and PTLS in clinical settings.
RAI1 and weight
RAI1 haploinsufficiency has emerged as a monogenic model for obesity. Studies in Rai1 +/− mice reveal significant weight gain compared to wild-type (WT) mice, with weight differences apparent by 20 weeks of age under standard feeding conditions (Ref. Reference Burns, Schmidt, Williams, Kim, Girirajan and Elsea60). Both male and female Rai1 +/− mice exhibited sustained weight gain into adulthood, approximately doubling the weight gain rate of WT mice (Ref. Reference Burns, Schmidt, Williams, Kim, Girirajan and Elsea60). This was accompanied by hyperphagia, impaired satiety and increased abdominal and subcutaneous fat distribution (Ref. Reference Burns, Schmidt, Williams, Kim, Girirajan and Elsea60). Interestingly, metabolic syndrome was not initially observed in these mice, as levels of adiponectin, insulin, glucose, total cholesterol and triglycerides were comparable to WT controls (Ref. Reference Burns, Schmidt, Williams, Kim, Girirajan and Elsea60). However, later studies on Rai1 +/− mice reported metabolic alterations, suggesting that genetic background may influence metabolic phenotypes (Ref. Reference Javed, Chang, Cho, Lee, Chang, Haque, Lin and Huang77). In contrast, Df(11)17/+ mice – carrying a broader chromosomal deletion – exhibited metabolic syndrome characteristics, including elevated fat content, reduced high-density lipoprotein levels and decreased insulin sensitivity (Ref. Reference Lacaria, Saha, Potocki, Bi, Yan, Girirajan, Burns, Elsea, Walz, Chan, Lupski and Gu55). This discrepancy suggests that neighbouring genes near Rai1 may modulate metabolic phenotypes, complicating the relationship between RAI1 and obesity (Refs Reference Lacaria, Saha, Potocki, Bi, Yan, Girirajan, Burns, Elsea, Walz, Chan, Lupski and Gu55, Reference Burns, Schmidt, Williams, Kim, Girirajan and Elsea60).
Mechanistic insights were provided by RNA sequencing of the hypothalamus in Rai1 +/− mice, which revealed reduced expression of BDNF. Chromatin immunoprecipitation (ChIP)-Chip and luciferase reporter assays demonstrated that RAI1 regulates BDNF transcription via an intronic enhancer (Ref. Reference Burns, Schmidt, Williams, Kim, Girirajan and Elsea60). Javed et al. (Ref. Reference Javed, Lee, Xu and Huang58) further advanced understanding through an inducible Rai1 knockout mouse model, allowing targeted deletion of Rai1 postnatally. Deleting Rai1 at 3 or 8 weeks of age did not affect neurobehavioural functions but induced hyperphagia and obesity (Ref. Reference Javed, Lee, Xu and Huang58). Notably, selective overexpression of BDNF – either murine or human – in the paraventricular or ventromedial hypothalamic nuclei successfully reversed weight gain and hyperphagia in Rai1 +/− mice (Ref. Reference Javed, Lee, Xu and Huang58). Building on these findings, researchers applied paraventricular nucleus-specific CRISPR activation using recombinant adeno-associated virus during early adolescence (3–4 weeks of age) in Rai1 +/− mice (Ref. Reference Chang, Lee, Javed, Haque, Chang, Lin, Oram and Huang59). This therapy boosted endogenous Rai1 expression, delayed obesity onset and partially alleviated hyperphagia (Ref. Reference Chang, Lee, Javed, Haque, Chang, Lin, Oram and Huang59). Javed et al. (Ref. Reference Javed, Chang, Cho, Lee, Chang, Haque, Lin and Huang77) further refined our understanding of RAI1’s role in regulating body weight, revealing its impact on hypothalamic BDNF-producing neurons, particularly those in the paraventricular nucleus. The study demonstrated that loss of Rai1 from these BDNF-producing neurons induces obesity in mice, with alterations in neuronal excitability and energy homeostasis (Ref. Reference Javed, Chang, Cho, Lee, Chang, Haque, Lin and Huang77). Interestingly, the researchers identified LM22A-4, a small molecule that activates neurotrophin signalling, which delayed obesity onset and partially improved metabolic phenotypes in Rai1+/− mice (Ref. Reference Javed, Chang, Cho, Lee, Chang, Haque, Lin and Huang77).
Evidences above underscore the pivotal role of RAI1 in hypothalamic energy balance regulation via postnatal BDNF expression. Early interventions targeting RAI1, particularly before adolescence, hold promise for mitigating SMS-related obesity and associated behavioural phenotypes (Refs Reference Huang, Wang, Allen, Klope, Hu, Shamloo and Luo13, Reference Chang, Lee, Javed, Haque, Chang, Lin, Oram and Huang59, Reference Javed, Lee, Xu and Huang58).
RAI1 and sleep disturbance
Sleep disturbances are a hallmark of SMS and PTLS, but they are significantly more prevalent in SMS patients (92.8%) compared with PTLS patients (39.1%; p < 0.001) (Table 1). Both patient data and animal models highlight the critical role of RAI1 in circadian rhythm regulation, with RAI1 haploinsufficiency appearing more disruptive than overexpression. Animal studies reveal that Df(11)17/+ mice and Rai1 +/− mice exhibit marked circadian abnormalities, primarily characterised by a shortened period length (Refs Reference Walz, Spencer, Kaasik, Lee, Lupski and Paylor52, Reference Williams, Zies, Mullegama, Grotewiel and Elsea61, Reference Lacaria, Gu and Lupski62, Reference Diessler, Kostic, Arsenijevic, Kawasaki and Franken63). In contrast, Dp(11)17/+ mice showed either normal or milder circadian rhythm disturbances (Refs Reference Mullegama, Alaimo, Fountain, Burns, Balog, Chen and Elsea12, Reference Walz, Spencer, Kaasik, Lee, Lupski and Paylor52, Reference Lacaria, Gu and Lupski62). These observations support a dose-dependent effect of RAI1 on circadian behaviour.
Mechanistic insights come from studies showing that RAI1 knockdown alters the expression of key circadian components, including CLOCK and other critical genes in the circadian feedback loop (Ref. Reference Williams, Zies, Mullegama, Grotewiel and Elsea61). This disruption has been observed in human cell lines (HEK 293T, U2OS-B), fibroblasts derived from SMS patients and the hypothalamus of Rai1 +/− mice (Ref. Reference Williams, Zies, Mullegama, Grotewiel and Elsea61). ChIP-Chip and luciferase data confirmed RAI1 as a positive regulator of CLOCK and an integral part of the circadian transcription loop (Ref. Reference Williams, Zies, Mullegama, Grotewiel and Elsea61). Interestingly, Dp(11)17/+ mice also exhibit dysregulation of circadian genes (CLOCK, Arntl, Per1–3 and Cry1/2) in the hypothalamus, indicating that RAI1 overexpression can also impair circadian function (Ref. Reference Mullegama, Alaimo, Fountain, Burns, Balog, Chen and Elsea12). This suggests that both insufficient and excessive RAI1 disrupt the molecular clock’s transcriptional regulation. In addition to downregulating CLOCK expression, Diessler et al. (Ref. Reference Diessler, Kostic, Arsenijevic, Kawasaki and Franken63) highlighted the involvement of Rai1 in processing non-visual light information, noting that Rai1 haploinsufficiency increased the sensitivity of Rai1 +/− mice to light stimuli. This discovery offered a new mechanism to understand sleep disturbances in SMS patients.
The cumulative evidence supports the core role of Rai1 in the central pathways of the sleep-wake cycle and circadian rhythm, indicating that both Rai1 deficiency and overexpression disrupt the transcriptional regulation of the molecular clock.
RAI1 and seizures
Seizures or abnormal electroencephalogram patterns were observed in a subset of SMS patients (27.7%). The work of Chang et al. (Ref. Reference Chang, Kowalczyk, Fogerson, Lee, Haque, Adams, Wang, DeNardo, Tessier-Lavigne, Huguenard, Luo and Huang14) provided insights into the underlying mechanisms, indicating that the absence of Rai1 enhanced hippocampal excitability and promoted epileptogenesis in SMS models. Specifically, the researchers employed whole-brain Rai1 homozygous knockout mice, combined with in vivo and ex vivo electrophysiological recordings, as well as in vivo brain structural and metabolic imaging, to uncover the involvement of Rai1 in modulating neuronal excitability (Ref. Reference Chang, Kowalczyk, Fogerson, Lee, Haque, Adams, Wang, DeNardo, Tessier-Lavigne, Huguenard, Luo and Huang14). In this context, hippocampal dentate gyrus granule cells exhibited heightened intrinsic excitability and augmented glutamatergic synaptic transmission (Ref. Reference Chang, Kowalczyk, Fogerson, Lee, Haque, Adams, Wang, DeNardo, Tessier-Lavigne, Huguenard, Luo and Huang14). In addition, insights from Garay et al. (Ref. Reference Garay, Chen, Tsukahara, Rodríguez Díaz, Kohen, Althaus, Wallner, Giger, Jones, Sutton and Iwase78) shed light on the synaptic Rai1-mediated gene transcription regulation, emphasising the unique role of Rai1 in homeostatic synaptic plasticity. The reduced expression of Rai1 induced functional changes in excitatory synaptic function, potentially contributing to alterations in neuronal excitability and the occurrence of seizures.
Discussion
This review provides a systematic examination of RAI1 and its involvement in a range of neuropsychiatric diseases, with particular relevance to paediatric conditions. In SMS and PTLS, RAI1 mutations lead to clear neurological and behavioural challenges, including developmental delays, sleep disturbances and cognitive impairments. These findings underscore RAI1’s critical involvement in early brain development, which is especially important for paediatric healthcare providers. While some conditions, such as ASD, typically manifest in early childhood, others, such as schizophrenia and bipolar disorder, often emerge in late adolescence or early adulthood. In contrast, SCA is generally classified as a group of adult-onset neurodegenerative disorders. The involvement of RAI1 in these conditions underscores its crucial role in maintaining neuropsychiatric health across different life stages.
Both reduced and overexpressed RAI1 can lead to overlapping neuropsychiatric phenotypes. However, as mentioned above, the occurrence and severity of these overlapping symptoms differ between SMS and PTLS patients. The summarised evidences from rare case reports emphasise the dose sensitivity of RAI1 during early embryonic development, highlighting the necessity for precise control of RAI1 dosage throughout development to ensure appropriate neurobehavioural functions in adulthood. Although recent studies have linked RAI1 to amyotrophic lateral sclerosis (Ref. Reference Yang, Li, Wei, Pang, Cheng, Huang, Lin, Xiao, Jiang, Wang and Shang79) and Parkinson’s disease (Ref. Reference Stern, Hussein, Cordeiro, Sadis, Garin-Shkolnik, Spiegel, Cohen, Harari, Schlesinger and Stern80), as well as benign adult familial myoclonic epilepsy type 8 (Ref. Reference Yeetong, Dembélé, Pongpanich, Cissé, Srichomthong, Maiga, Dembélé, Assawapitaksakul, Bamba, Yalcouyé, Diarra, Mefoung, Rakwongkhachon, Traoré, Tongkobpetch, Fischbeck, Gahl, Guinto, Shotelersuk and Landouré81), its role in the mature nervous system remains poorly understood. Notably, current research has primarily focused on RAI1’s dosage sensitivity – that is, the effects of insufficient or excessive expression – while largely overlooking its intrinsic biochemical properties. For instance, nonsense and frameshift mutations occurring upstream of the nuclear localisation signal result in truncated proteins, which not only lead to RAI1 haploinsufficiency but also raise the question of whether the mislocalised N-terminal fragment, retained in the cytoplasm, exerts dominant-negative or toxic effects. Furthermore, given that RAI1 exceeds 200 kDa in molecular weight and contains intrinsically disordered regions, it may exhibit a propensity for aggregation. This raises a critical yet largely unexplored question: Does RAI1 duplication not only increase expression but also promote aberrant self-aggregation? If so, what are the functional and pathological consequences of such aggregation? Addressing these fundamental questions will be essential for advancing our understanding of RAI1’s molecular properties and its broader implications in neurodevelopmental and neurodegenerative disorders.
In addition to chromosomal structural variations and point mutations, the RAI1 gene is also susceptible to CAG repeat expansions (Ref. Reference Elsea and Williams1). Normal RAI1 CAG repeats range from 10 to 19 in the general population, with 13 or 14 repeats being most common (Refs Reference Bi, Saifi, Girirajan, Shi, Szomju, Firth, Magenis, Potocki, Elsea and Lupski22, Reference Ivković, Zamurović, Jovanović, Dobričić, Damjanović, Savić-Pavićević and Romac82, Reference Tan, Tan, Chua, Lee, Ng, Ch’ng, Choo and Chen83). Indeed, expansion of CAG repeats is associated not only with SCA and Huntington’s disease but also with other neurodegenerative conditions (Ref. Reference Rosas, Martínez, Clarimón, Lleó, Illán-Gala, Dols-Icardo, Borroni, Almeida, van der Zee, Van Broeckhoven, Bruni, Anfossi, Bernardi, Maletta, Serpente, Galimberti, Scarpini, Rossi, Caroppo, Benussi, Ghidoni, Binetti, Nacmias, Sorbi, Piaceri, Bagnoli, Antonell, Sánchez-Valle, De la Casa-Fages, Grandas, Diez-Fairen, Pastor, Ferrari, Álvarez and Menéndez-González84). The finding that CAG repeat length in RAI1 modifies the age of onset in SCA patients aligns with the broader theory that (CAG)n repeat expansions influence disease onset (Ref. Reference Tezenas du Montcel, Durr, Bauer, Figueroa, Ichikawa, Brussino, Forlani, Rakowicz, Schols, Mariotti, van de Warrenburg, Orsi, Giunti, Filla, Szymanski, Klockgether, Berciano, Pandolfo, Boesch, Melegh, Timmann, Mandich, Camuzat, Goto, Ashizawa, Cazeneuve, Tsuji, Pulst, Brusco, Riess, Brice and Stevanin66). However, these findings need validation through larger and more diverse population studies, and further research is necessary to confirm the impacts at the cellular level.
Evidence from genome-wide association studies (Ref. Reference Trost, Thiruvahindrapuram, Chan, Engchuan, Higginbotham, Howe, Loureiro, Reuter, Roshandel, Whitney, Zarrei, Bookman, Somerville, Shaath, Abdi, Aliyev, Patel, Nalpathamkalam, Pellecchia, Hamdan, Kaur, Wang, MacDonald, Wei, Sung, Lamoureux, Hoang, Selvanayagam, Deflaux, Geng, Ghaffari, Bates, Young, Ding, Shum, D’Abate, Bradley, Rutherford, Aguda, Apresto, Chen, Desai, Du, MLY, Pullenayegum, Samler, Wang, Ho, Paton, Pereira, Herbrick, Wintle, Fuerth, Noppornpitak, Ward, Magee, Al Baz, Kajendirarajah, Kapadia, Vlasblom, Valluri, Green, Seifer, Quirbach, Rennie, Kelley, Masjedi, Lord, Szego, Zawati, Lang, Strug, Marshall, Costain, Calli, Iaboni, Yusuf, Ambrozewicz, Gallagher, Amaral, Brian, Elsabbagh, Georgiades, Messinger, Ozonoff, Sebat, Sjaarda, Smith, Szatmari, Zwaigenbaum, Kushki, Frazier, JAS, Fakhro, Fernandez, MES, Weksberg, Fiume, RKC, Anagnostou, Sondheimer, Glazer, Hartley and Scherer9) and post-mortem brain analyses (Ref. Reference Haybaeck, Postruznik, Miller, Dulay, Llenos and Weis10) strongly implicates RAI1 in various psychiatric disorders, although direct research on its role in ASD, schizophrenia, bipolar disorder and major depression remains limited. Moreover, both SMS and PTLS patients present with significant psychiatric symptoms, such as autistic features, schizophrenia, compulsive behaviour, aggressive behaviour, self-injurious behaviour, depression and anxiety, suggesting that both reduced and elevated RAI1 expression contribute to these phenotypes. In SMS and PTLS, these symptoms primarily stem from aberrant RAI1 expression, whereas in other psychiatric disorders, RAI1 likely acts within broader genetic networks influencing disease susceptibility and clinical presentation. Of course, further studies are needed to clarify the specific and shared roles of RAI1 in psychiatric conditions and the underlying mechanisms.
RAI1 is a highly conserved dosage-sensitive gene across species. Most fundamental studies have primarily focused on mouse models with altered gene expression to mimic SMS and PTLS, providing key mechanistic insights. However, there is currently no available evidence from other model organisms, such as zebrafish or Drosophila, highlighting the need for further investigation. As a transcription factor, RAI1 regulates diverse biological processes, including neuronal differentiation, cell growth, skeletal development, lipid biosynthesis, glucose metabolism, behaviour and circadian rhythms (Ref. Reference Girirajan, Truong, Blanchard and Elsea85). In SMS, RAI1 haploinsufficiency in the hypothalamus reduces BDNF and CLOCK expression, contributing to obesity and sleep disturbances, respectively (Refs Reference Mullegama, Alaimo, Fountain, Burns, Balog, Chen and Elsea12, Reference Javed, Lee, Xu and Huang58). Additionally, studies have linked Rai1 deficiency in hippocampal and glutamatergic neurons to heightened excitability and enhanced excitatory synaptic transmission, underlying epilepsy phenotypes (Ref. Reference Chang, Kowalczyk, Fogerson, Lee, Haque, Adams, Wang, DeNardo, Tessier-Lavigne, Huguenard, Luo and Huang14). Mouse models have proven particularly effective for studying weight abnormalities, sleep disturbances and epilepsy, as these phenotypes are relatively easy to replicate and monitor. These investigations underscore the critical role of precisely regulated RAI1 expression and transcriptional activity in maintaining normal brain function. Moreover, recent studies have further characterised the neuroanatomical distribution and molecular interactions of RAI1 in both mice and primates (marmosets) (Ref. Reference Chang, Lee, Haque, Chang, Javed, Lin, Cho, Abramovitz, Chin, Khamis, Raja, Murai and Huang86). Overall, the specific mechanisms by which RAI1 contributes to a broader spectrum of neurological and psychiatric disorders require further exploration.
Several limitations of this study should be acknowledged. First, our review only included studies in the English language published in the PubMed and EMBASE databases, potentially missing some relevant published studies. Second, in the clinical research review section, the majority of reported SMS and PTLS patients were from the United States and European countries, limiting the representativeness of the samples and possibly not accounting for potential genetic and phenotypic differences in patients from other regions. Future studies with larger and more diverse cohorts are necessary to systematically quantify RAI1 genotype–phenotype associations. Third, as illustrated in Table 1, SMS and PTLS patients manifested a diverse array of phenotypes, including but not limited to craniofacial and skeletal development, cognition, motor skills, sensory functions, psychiatric behaviour, cardiovascular system and urinary system development. However, in the basic research review section, we primarily focused on summarising the currently more extensive and in-depth mechanistic studies, specifically those related to the mechanisms of RAI1 in regulating weight, circadian rhythm and epilepsy.
Conclusions
This review highlights the role of RAI1 in a range of neuropsychiatric diseases, including SMS, PTLS, SCA, ASD, schizophrenia, bipolar disorder and major depression. Starting with the summary of SMS and PTLS cases and their corresponding mouse model phenotypic mechanisms, we gained insights into the pivotal role of RAI1 in neurodevelopment and functional maintenance, emphasising the importance of precise gene dosage. Future research endeavours should address several key aspects: (1) further exploring the mechanisms behind the diverse phenotypes observed in SMS and PTLS, (2) validating RAI1’s role in various neurological and psychiatric conditions and (3) expanding our understanding of its regulatory network in the adult brain. A deeper understanding of RAI1’s functions will not only enhance knowledge of these disorders but also pave the way for potential therapeutic interventions.
Supplementary material
The supplementary material for this article can be found at http://doi.org/10.1017/erm.2025.12.
Acknowledgements
We express our gratitude to all the original research studies whose valuable contributions have enriched this comprehensive review.
Data availability statement
All data generated or analysed during this study are included in this published article (and its Supplementary Materials).
Funding statement
This study was supported by the National Natural Science Foundation of China (Grant No. 82371430), the National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University (Grant No. Z2024JC004), and the Science and Technology Bureau Fund of Sichuan Province of China (Grant No. 2023YFQ0098).
Competing interests
The authors declare no competing interests.






 Open access
Open access