


Introduction
DNA methylation (DNAm)-derived epigenome-wide scores have emerged as leading biomarkers of biological age and death (e.g., Horvath, Reference Horvath2013; Lu et al., Reference Lu, Quach, Wilson, Reiner, Aviv, Raj and Horvath2019) and are referred to as “DNAm age.” Estimates of DNAm age may differ from chronological age, such that some individuals evidence advanced DNAm age relative to chronological age. Advanced DNAm age has been linked to age-related disease and adverse health outcomes, including metabolic syndrome, inflammation, neuropathology, and mortality (Chen et al., Reference Chen, Marioni, Colicino, Peters, Ward-Caviness, Tsai and Horvath2016; Christiansen et al., Reference Christiansen, Lenart, Tan, Vaupel, Aviv, Mcgue and Christensen2016; Gassen, Chrousos, Binder, & Zannas, Reference Gassen, Chrousos, Binder and Zannas2017; Hillary et al., Reference Hillary, Stevenson, Cox, McCartney, Harris, Seeboth and Marioni2021; Levine et al., Reference Levine, Hosgood, Chen, Absher, Assimes and Horvath2015; Marioni et al., Reference Marioni, Harris, Shah, McRae, von Zglinicki, Martin-Ruiz and Deary2016, Reference Marioni, Shah, McRae, Chen, Colicino, Harris and Deary2015; Meier, Mitchell, Karadimas, & Faul, Reference Meier, Mitchell, Karadimas and Faul2023; Miller & Sadeh, Reference Miller and Sadeh2014; Reed, Carroll, Marsland, & Manuck, Reference Reed, Carroll, Marsland and Manuck2022), highlighting the applied significance of DNAm age as a biomarker of biological aging and its potential for identifying individuals at risk for early onset of age-related diseases.
Two of the DNAm age algorithms that have received the most attention to date are the Horvath (Horvath, Reference Horvath2013) marker of biological age and the Lu et al. (Reference Lu, Quach, Wilson, Reiner, Aviv, Raj and Horvath2019) index of time until death, referred to as “GrimAge.” Both algorithms were trained through machine learning approaches to select a set of CpG sites from the epigenome that optimize prediction. The regression coefficients of the selected CpG sites are then used as weights in novel datasets to calculate the respective DNAm age estimates. Age-adjusted DNAm age is commonly defined as the residuals from regressing DNAm age on chronological age where negative values represent slowed epigenetic age and positive values represent advanced epigenetic age relative to chronological age.
Several studies have examined psychiatric diagnoses and symptoms in association with advanced DNAm age in blood using cross-sectional methods, including studies of depression (Liu et al., Reference Liu, Wang, Hui, Goldberg, Smith, Shah and Vaccarino2022) and alcohol use disorders (Rosen et al., Reference Rosen, Robertson, Hlady, Muench, Lee, Philibert and Lohoff2018). Posttraumatic stress disorder (PTSD) has received particular attention in this regard, with numerous individual studies (Jovanovic et al., Reference Jovanovic, Vance, Cross, Knight, Kilaru, Michopoulos and Smith2017; Katrinli et al., Reference Katrinli, Stevens, Wani, Lori, Kilaru, van Rooij and Smith2020; Roberts et al., Reference Roberts, Koenen, Chen, Gilsanz, Mason, Prescott and Kubzansky2017; Wolf et al., Reference Wolf, Logue, Hayes, Sadeh, Schichman, Stone and Miller2016; Wolf, Logue et al., Reference Wolf, Logue, Stoop, Schichman, Stone, Sadeh and Miller2018) and a meta-analysis (Wolf, Maniates et al., Reference Wolf, Maniates, Nugent, Maihofer, Armstrong, Ratanatharathorn and Logue2018) providing support for cross-sectional associations between PTSD and advanced DNAm age. Specifically, in the meta-analysis which was based on cross-sectional data from 9 cohorts contributing to the Psychiatric Genomics Consortium (5 of which contributed longitudinal data to these analyses), we previously found that the lifetime PTSD severity, but not PTSD diagnosis, and childhood trauma exposure (when measured with a consistent trauma measure across studies) were associated with increased epigenetic age (Wolf, Maniates et al., Reference Wolf, Maniates, Nugent, Maihofer, Armstrong, Ratanatharathorn and Logue2018). This is consistent with the hypothesis that the cumulative burden of PTSD symptom severity across the lifespan, including the chronic toll of physiological reactivity, poor sleep, anger, and arousal, may be most relevant for understanding risk for accelerated aging. Some studies have further linked PTSD-related advanced DNAm age with biomarkers of inflammation, metabolic pathology, and neuropathology (Morrison et al., Reference Morrison, Logue, Guetta, Maniates, Stone, Schichman and Wolf2019; Wolf et al., Reference Wolf, Miller, Hawn, Zhao, Wallander, McCormick and Logue2023), suggesting that individuals with PTSD may be at greater risk for developing early onset of these conditions. However, cross-sectional designs cannot address questions concerning the directionality of associations between PTSD and advanced DNAm age or the temporal stability of estimates of advanced epigenetic age over time, nor can they track how PTSD-related changes in epigenetic age accumulate and contribute to long-term health outcomes. Therefore, studying the longitudinal relationship between epigenetic aging and PTSD is crucial for understanding how trauma-related disorders contribute to premature aging and long-term health decline over time, as well as for developing targeted interventions that are matched to the pathophysiology that contributes to early onset of these health conditions.
To date, only a few studies have examined the relationship between PTSD and advanced DNAm age longitudinally, and the approach to modeling changes in DNAm age estimates over time has varied across studies, as have results. In a longitudinal cohort of 96 male Dutch soldiers assessed prior to war zone deployment and six months post-deployment, intervening combat trauma was found to be associated with an increase in raw (i.e., not accounting for chronological age) estimates of Horvath DNAm age over time (Boks et al., Reference Boks, Mierlo, Rutten, Radstake, De Witte, Geuze and Vermetten2015). However, increased PTSD symptoms post-deployment were negatively associated with the change in raw DNAm age between time points (Boks et al., Reference Boks, Mierlo, Rutten, Radstake, De Witte, Geuze and Vermetten2015), suggesting potential differential effects of trauma exposure versus PTSD.
Using an alternative analytic design, a study of 40 paramedicine students assessed twice (pre- and post-work-related trauma exposure) found that baseline Horvath DNAm age residuals were positively associated with PTSD severity at follow-up 12 months later (about 1–2 months following trauma exposure; Mehta et al., Reference Mehta, Bruenig, Pierce, Sathyanarayanan, Stringfellow, Miller and Shakespeare-Finch2022). However, all participants reported trauma histories at baseline, raising the possibility that baseline DNAm age was advanced due to this and/or associated pre-existing psychiatric symptoms. Consistent with this possibility, the same study found that baseline PTSD severity was associated with greater Horvath and GrimAge DNAm age residuals at follow-up (Mehta et al., Reference Mehta, Bruenig, Pierce, Sathyanarayanan, Stringfellow, Miller and Shakespeare-Finch2022). Most critically, the analyses did not account for baseline DNAm age acceleration, masking the extent to which DNAm age changed from baseline to follow-up. Another study (Yang et al., Reference Yang, Wu, Verhoeven, Gautam, Reus, Kang and Wolkowitz2021) modeled change in DNAm age estimates through the use of correlated change scores between GrimAge residuals and PTSD symptom severity scores among trauma-exposed male military Veterans assessed twice. Among those who had PTSD at baseline, change in PTSD severity across 3 years was correlated positively with change in GrimAge residuals, but this was based on just 26 participants. Concerns about small sample size (and associated limitations to statistical power and representativeness) apply to all the longitudinal studies of PTSD and epigenetic aging to date.
Other studies have addressed the challenges of modeling the relationships between DNAm age and time between assessments by examining the rate of change in raw DNAm age estimates relative to the time between assessments. One study (Sumner et al., Reference Sumner, Gao, Gambazza, Dye, Colich, Baccarelli and McLaughlin2023) followed a group of 171 children and adolescents over 2 years and found that the rate of change in DNAm age was greater (more positive) among those who experienced more negative impact from intervening stressful life events. Two other studies of the rate of change among Veteran cohorts with chronic PTSD found that baseline PTSD symptom severity and diagnosis predicted an increased pace of Horvath DNAm age over the course of approximately 2 (Wolf et al., Reference Wolf, Logue, Morrison, Wilcox, Stone, Schichman and Miller2019) and 5.5 (Hawn et al., Reference Hawn, Zhao, Miller, Wallander, Govan, Stone and Wolf2023) years, respectively. Collectively, these studies, with sample sizes ranging from 26 to 179, highlight the challenges in modeling change over time in DNAm age and raise the possibility that different analytic strategies may be necessary to best address questions concerning how new-onset PTSD diagnoses versus chronic symptoms relate to changes in DNAm age over time.
Aims and hypotheses
This study sought to investigate the health correlates of PTSD in terms of changes in epigenetic aging over time. We examined the relationship between PTSD and future changes in epigenetic age acceleration over time using meta-analysis of longitudinal data from seven cohorts contributing to the Psychiatric Genomics Consortium (PGC) PTSD Epigenetics Workgroup (Ratanatharathorn et al., Reference Ratanatharathorn, Boks, Maihofer, Aiello, Amstadter, Ashley-Koch and Smith2017). The varied methodological structures of these datasets allowed us to ask multiple questions about the relationship between PTSD and epigenetic age. Specifically, our first aim was to evaluate if the association between Time 1 (T1) and Time 2 (T2) DNAm age residuals was altered by the development of new-onset PTSD between two time points. We expected that the association between DNAm age residuals across two time points would be more positive among those with new-onset PTSD at follow-up. Our second aim was to examine whether the association between DNAm age residuals at two time points varied as a function of change in PTSD symptom severity (T2–T1). We also hypothesized that the association between DNAm age residuals at two time points would be stronger as the change in PTSD symptom severity increased. Both hypotheses were tested by modeling the association between DNAm age residuals at T2 with the T1 DNAm age residuals × new-onset PTSD and T1 DNAm age residuals × change in PTSD symptom severity interaction terms, respectively, while adjusting for their main effects and other covariates. These aims were addressed in a total of 1,367 individuals derived from 7 cohorts who were each assessed for DNAm twice. The cohorts spanned both civilian and military samples and included research methods that were focused on pre/post military deployment or pre/post trauma exposure, and those focused on chronic PTSD symptoms. The former design allowed for the examination of new-onset PTSD diagnoses over time while the latter design allowed us to examine changes in PTSD symptom severity over time in association with changes in DNAm age residuals. We were unable to examine the changing rate of epigenetic aging over time (e.g., Wolf et al., Reference Wolf, Logue, Morrison, Wilcox, Stone, Schichman and Miller2019) due to the structure of the contributing datasets, which included many cohorts defined by pre/post trauma exposure.
Analyses focused on Horvath age (Horvath, Reference Horvath2013) and GrimAge (Lu et al., Reference Lu, Quach, Wilson, Reiner, Aviv, Raj and Horvath2019) as they represent the most widely used and robust metrics of biological age and time to death, respectively (sometimes referred to as first- versus second-generation clocks). In addition, these are the only DNAm age indices previously associated with PTSD in small longitudinal studies (Boks et al., Reference Boks, Mierlo, Rutten, Radstake, De Witte, Geuze and Vermetten2015; Hawn et al., Reference Hawn, Zhao, Miller, Wallander, Govan, Stone and Wolf2023; Mehta et al., Reference Mehta, Bruenig, Pierce, Sathyanarayanan, Stringfellow, Miller and Shakespeare-Finch2022; Sumner et al., Reference Sumner, Gao, Gambazza, Dye, Colich, Baccarelli and McLaughlin2023; Wolf et al., Reference Wolf, Miller, Hawn, Zhao, Wallander, McCormick and Logue2023; Yang et al., Reference Yang, Wu, Verhoeven, Gautam, Reus, Kang and Wolkowitz2021). We also sought to limit the number of tests to reduce the burden of multiple testing corrections and thus chose to limit analyses to the strongest epigenetic age metrics with prior evidence of longitudinal associations with PTSD.
Methods and materials
Participating studies
Seven participating cohorts were included in the meta-analysis. The mean time between assessments within each study ranged from 5.7 months to 5.6 years. The cohorts included: (1) The Longitudinal National Center for PTSD (NCPTSD) cohort (Wolf et al., Reference Wolf, Miller, Hawn, Zhao, Wallander, McCormick and Logue2023), a study of trauma-exposed Veterans (many with chronic PTSD), who were assessed twice, an average of 5.6 years apart; (2) the Translational Research Center for TBI and Stress Disorders (TRACTS) cohort (McGlinchey, Milberg, Fonda, & Fortier, Reference McGlinchey, Milberg, Fonda and Fortier2017), which consisted of post-9/11 Veterans (many with chronic PTSD) who completed two assessments an average of 1.9 years apart; (3) the Army Study to Assess Risk and Resilience in Servicemembers (Army STARRS) (Ursano et al., Reference Ursano, Colpe, Heeringa, Kessler, Sehoenbaum and Stein2014), a study of military service members assessed pre- and post-deployment to Afghanistan over an average of 9.6 months; (4) the Marine Resiliency Study (MRS) cohort (Baker et al., Reference Baker, Nash, Litz, Geyer, Risbrough, Nievergelt and Webb-Murphy2012; Nievergelt et al., Reference Nievergelt, Maihofer, Mustapic, Yurgil, Schork, Miller and Baker2015) of male US Marines assessed pre- and post-deployment to Iraq or Afghanistan over a mean interval of 12.4 months; (5) the Prospective Research in Stress-related Military Operations (PRISMO) cohort (Eekhout, Reijnen, Vermetten, & Geuze, Reference Eekhout, Reijnen, Vermetten and Geuze2016; Reijnen, Rademaker, Vermetten, & Geuze, Reference Reijnen, Rademaker, Vermetten and Geuze2015) of Dutch soldiers who were assessed pre- and post-deployment to Afghanistan over an average of 14.4 months; (6) the Detroit Neighborhood Health Study (DNHS) cohort (Goldmann et al., Reference Goldmann, Aiello, Uddin, Delva, Koenen, Gant and Galea2011; Uddin et al., Reference Uddin, Aiello, Wildman, Koenen, Pawelec, De Los Santos and Galea2010) which consisted of trauma-exposed Detroit residents (some with chronic psychiatric symptoms) who completed two assessments an average of 16.3 months apart; and (7) the Advancing Understanding of Recovery after Trauma (AURORA) Study (McLean et al., Reference McLean, Ressler, Koenen, Neylan, Germine, Jovanovic and Kessler2020), which included individuals evaluated at the emergency department following trauma exposure who were reassessed an average of 5.7 months after emergency department treatment. Table 1 lists the demographic and clinical characteristics of each cohort. Each study site obtained local IRB approval, and all participants provided written informed consent. The IRB of the VA Boston Healthcare System approved the meta-analyses of the summarized data. Individuals interested in obtaining access to the data should contact the principal investigator of each individual cohort to determine availability.
Table 1. Cohort characteristics
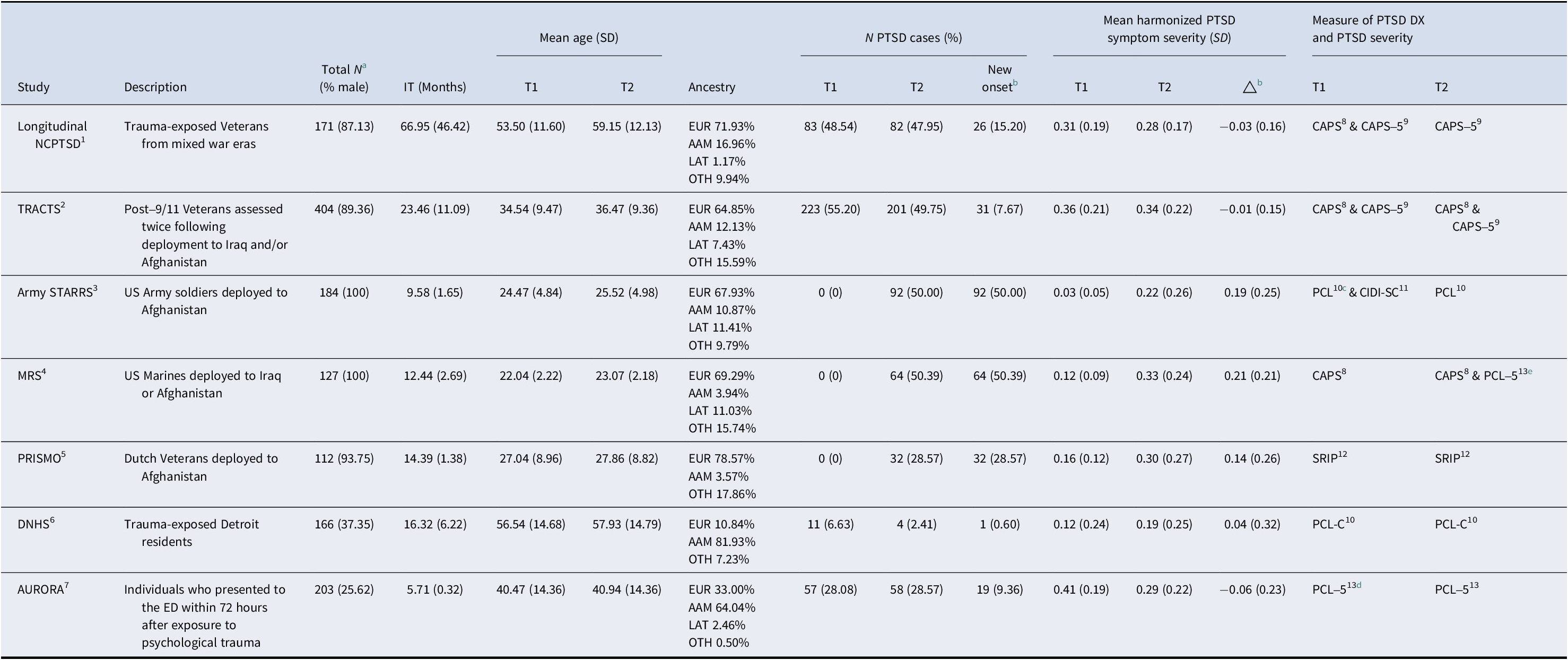
Note: Superscripted numbers refer to the references for each cohort and PTSD measure: 1. Wolf et al. (Reference Wolf, Miller, Hawn, Zhao, Wallander, McCormick and Logue2023); 2. McGlinchey et al. (Reference McGlinchey, Milberg, Fonda and Fortier2017); 3. Ursano et al. (Reference Ursano, Colpe, Heeringa, Kessler, Sehoenbaum and Stein2014); 4. Baker et al. (Reference Baker, Nash, Litz, Geyer, Risbrough, Nievergelt and Webb-Murphy2012); Nievergelt et al. (Reference Nievergelt, Maihofer, Mustapic, Yurgil, Schork, Miller and Baker2015); 5. Eekhout et al. (Reference Eekhout, Reijnen, Vermetten and Geuze2016); Reijnen et al. (Reference Reijnen, Rademaker, Vermetten and Geuze2015); 6. Goldmann et al. (Reference Goldmann, Aiello, Uddin, Delva, Koenen, Gant and Galea2011); Uddin et al. (Reference Uddin, Aiello, Wildman, Koenen, Pawelec, De Los Santos and Galea2010); 7. McLean et al. (Reference McLean, Ressler, Koenen, Neylan, Germine, Jovanovic and Kessler2020); 8. Blake et al. (Reference Blake, Weathers, Nagy, Kaloupek, Gusman, Charney and Keane1995); 9. Weathers et al. (Reference Weathers, Bovin, Lee, Sloan, Schnurr, Kaloupek and Marx2018); 10. Weathers et al. (Reference Weathers, Litz, Herman, Huska and Keane1993); 11. Kessler et al. (Reference Kessler, Santiago, Colpe, Dempsey, First, Heeringa and Ursano2013); 12. Hovens, Bramsen, and Van Der Ploeg (Reference Hovens, Bramsen and Van Der Ploeg2002); 13. Blevins et al. (Reference Blevins, Weathers, Davis, Witte and Domino2015).
Abbreviations: NCPTSD, The National Center for PTSD Study; TRACTS, The Translational Research Center for TBI and Stress Disorders Study; Army STARRS, The Army Study to Assess Risk and Resilience in Servicemembers; MRS, The Marine Resilience Study; PRISMO, The Prospective Research in Stress-related Military Operations; DNHS, The Detroit Neighborhood Health Study; AURORA, The Advancing Understanding of RecOvery afteR traumA Study; IT, Intervening time; PTSD, posttraumatic stress disorder; DX, diagnosis; T1, time 1; T2, time 2; ED, emergency department; EUR, European ancestry; AAM, African American ancestry; LAT, Latino ancestry; OTH, Other ancestries; PCL, PTSD Checklist for DSM-IV; PCL-5, PTSD Checklist for DSM-5; PCL-C, PTSD Checklist Civilian Version; DSM, Diagnostic and statistical manual of mental disorders; CIDI-SC, Composite International Diagnostic Interview screening scales; CAPS, Clinician-Administered PTSD Scale for DSM-IV; CAPS-5, Clinician-Administered PTSD Scale for DSM-5; SRIP, Self-Report Inventory for PTSD; △ = change in harmonized PTSD symptom severity (T2-T1); SD, standard deviation.
a Sample sizes for computing cross-sectional and longitudinal correlations among DNAm age, DNAm age residuals, and DNAm-based cell type proportions.
b Sample sizes for the new-onset PTSD diagnosis analysis and change in symptom severity analysis can be found in Figures 1 and 2. Compared to the total N, the sample size for the new-onset PTSD diagnosis analysis was reduced due to exclusion of PTSD cases at baseline and missing values in PTSD diagnosis and covariates. The sample size for the change in symptom severity analysis was reduced due to missing values in PTSD symptom severity and covariates.
c 6-item screening version of PCL.
d Abbreviated (six-item) civilian version of PCL-5.
e CAPS was used for determining PTSD DX and PCL-5 was used for assessing PTSD severity.
DNA and DNAm procedures
DNA was extracted from buffy coat from peripheral blood samples. DNAm was measured using Illumina Infinium EPIC BeadChip at two time points. DNAm data were processed following a quality control (QC) pipeline developed by the PGC PTSD Epigenetics Workgroup (Ratanatharathorn et al., Reference Ratanatharathorn, Boks, Maihofer, Aiello, Amstadter, Ashley-Koch and Smith2017; and its updated version at https://github.com/PGC-PTSD-EWAS/EPIC_QC). Details of QC procedures are included in the Supplementary Materials. DNAm is cell type-specific and thus DNAm levels may vary as a function of the composition of the cell types the DNA was extracted from. Given this, it is important to include proportional estimates of white blood cell types as covariates in analyses (as is standard in DNAm analyses). Proportions of six cell types (B cells, CD4+ T cells, CD8+ T cells, natural killer cells, monocytes, and neutrophils) were estimated directly from the DNAm data using a reference library of CpG sites aligned with sorted cells, as implemented in the Bioconductor package EpiDISH (Teschendorff, Breeze, Zheng, & Beck, Reference Teschendorff, Breeze, Zheng and Beck2017). Of these, B cells, CD4+ T cells, CD8+ T cells, natural killer cells, and monocytes were included as covariates in the analysis (neutrophils are excluded because the proportional nature of the estimates makes them colinear when all other cell types are included in the model). Because blood-based methylation is strongly influenced by smoking, we computed a DNAm-based smoking score based on 39 smoking-associated CpGs (Li et al., Reference Li, Wong, Bui, Nguyen, Joo, Stone and Hopper2018; Supplementary Material). As our outcome of interest is epigenetic age acceleration at T2, the DNAm smoking score from T2 was included as an additional covariate in a sensitivity analysis to determine if our findings were driven by the effects of concurrent smoking, which can co-occur with PTSD.
Genotyping was conducted on various arrays. Genotype data cleaning was completed at each site according to the procedures previously described in the original publications for each study (Table 1). Ancestry was determined based on genotype data (where available) using the pipeline developed by the PGC PTSD (Nievergelt et al., Reference Nievergelt, Maihofer, Klengel, Atkinson, Chen, Choi and Koenen2019). Ancestry-based principal components (PCs) were computed from 100,000 randomly selected common single nucleotide polymorphisms (SNPs) with minor allele frequency (MAF) > 0.05. When genotyping data were unavailable (e.g., DNHS), methylation probes within 1 base pair of SNPs for determining ancestry were used to generate a set of DNAm-based ancestral PCs as proxies for genotype-based PCs (Barfield et al., Reference Barfield, Almli, Kilaru, Smith, Mercer, Duncan and Conneely2014). As noted from our previous meta-analysis study, the DNAm-derived PCs are significantly correlated with genotype-based PCs (Ratanatharathorn et al., Reference Ratanatharathorn, Boks, Maihofer, Aiello, Amstadter, Ashley-Koch and Smith2017; Wolf, Maniates, et al., Reference Wolf, Maniates, Nugent, Maihofer, Armstrong, Ratanatharathorn and Logue2018). Either the first three genotype-based PCs 1–3 or, when genotype-based PCs were unavailable, the DNAm-based PCs 2–4, were used as covariates in the analyses to account for ancestry.
Measures
PTSD measures
Current PTSD diagnosis and symptom severity were assessed using various measures as listed in Table 1. Following a previous PGC publication (Sumner et al., Reference Sumner, Maihofer, Michopoulos, Rothbaum, Almli, Andreassen and Wolf2021), we harmonized PTSD symptom severity across these different measures by scaling the raw current PTSD severity score to a range from 0 to 1, representing the score as a percentage of the maximum possible score on each measure (i.e., 1 represents an individual having all symptoms at the most severe level and 0 indicates an individual having no symptoms). For this study, we examined both new-onset PTSD diagnosis (e.g., no PTSD at T1 and PTSD positive at T2 versus no PTSD at T1 and T2) and changes in PTSD symptom severity (T2–T1) using the harmonized PTSD score. For the new-onset PTSD diagnosis analyses, we only included individuals who were negative for PTSD at T1, which resulted in reductions in sample size in some of the studies that included participants with chronic PTSD (Table 1). For the change in PTSD severity analyses, we included all participants with PTSD symptom severity data across all cohorts, regardless of PTSD diagnostic status at baseline.
DNAm age indices
The Horvath DNAm age and GrimAge estimates were computed by uploading the DNAm data to Dr. Horvath’s website (https://dnamage.genetics.ucla.edu/) when permitted or by running the scripts supplied by Drs. Horvath and Lu if methylation data were not allowed to be uploaded per local regulations (Horvath, Reference Horvath2013; Lu et al., Reference Lu, Quach, Wilson, Reiner, Aviv, Raj and Horvath2019). Horvath’s algorithm has its own normalization and imputation step, so raw DNAm values were used as the input for the Horvath age calculation. Values from 353 probes were used to generate Horvath age (Horvath, Reference Horvath2013). However, 17 (4.8%) of the 353 CpGs are missing from the EPIC chip (Dhingra et al., Reference Dhingra, Kwee, Diaz-Sanchez, Devlin, Cascio, Hauser and Ward-Caviness2019). A small number of additional missing probes from each cohort were identified as summarized in the Supplementary Materials. GrimAge estimates were generated using normalized and imputed DNAm data. 30,084 probes were used as input for GrimAge calculation (Lu et al., Reference Lu, Quach, Wilson, Reiner, Aviv, Raj and Horvath2019).
Both Horvath age and GrimAge residuals were computed by saving the unstandardized residuals from a linear model regressing the raw DNAm age on chronological age. This was done for each cohort at two time points separately, so the age residuals have a mean of 0 for each cohort at each time point. An R script was developed by the first author, tested with co-authors, and then sent to the data analyst at each participating cohort site so that identical calculation and analytic approaches would be applied in each cohort. Summary statistics from each cohort were then meta-analyzed to combine the results across studies.
Statistical analyses
We first examined the Pearson correlations between Horvath age and GrimAge with chronological age at each corresponding time point. We then assessed the correlations among Horvath age, GrimAge, Horvath age residuals, and GrimAge residuals over time. Correlations among cell types over time and between the two DNAm age residuals and cell types were also evaluated. The correlation coefficients collected from each group were meta-analyzed using the metacor (Laliberté, Reference Laliberté2022) package in R.
We conducted two primary regression analyses. The first focused on examining how new-onset PTSD diagnosis at T2 (versus remaining negative for PTSD at both time points) alone and in interaction with T1 DNAm age residuals predicted T2 DNAm age residuals, covarying for T1 DNAm age residuals and covariates. The second analysis replaced new-onset PTSD diagnosis with change in PTSD symptom severity over time. In both analyses, we first performed a regression model without the interaction term to capture the main effects of all predictors (all main effect coefficients reported are from this initial model) and then added the interaction term into the model. The interaction term examined the extent to which the association between T1 DNAm age residuals and T2 DNAm age residuals differed as a function of change in PTSD (diagnosis or severity). A positive interaction term would indicate T2 DNAm age residuals become more extreme than what would be predicted from T1 DNAm age residuals alone, as a function of the moderating effect of changes in PTSD status. This more extreme alteration could occur at both ends of the DNAm age residuals. In the primary linear regression models predicting T2 age residuals, the predictors in the model were DNAm age residuals at T1, new-onset PTSD diagnosis at T2 (or separately, change in PTSD symptom severity), the interaction between T1 DNAm age residuals and new-onset PTSD diagnosis (or change in PTSD severity), and the following covariates: sex (excluded if there was no variability in sex in a given sample), three ancestry PCs, and five cell type proportion estimates at T2. Significant associations were further examined in sensitivity analyses, including DNAm-based smoking scores at T2 as an additional covariate. Follow-up models evaluated the association between childhood trauma (predating T1) and changes in epigenetic age over time (Supplementary Materials).
Meta-analysis of each unstandardized parameter estimate in the regression models (except ancestry PCs) was conducted in an inverse variance-weighted random-effects model using the metafor package in R (Viechtbauer, Reference Viechtbauer2010). Results for each term were corrected for multiple testing via the false discovery rate (FDR) adjustment (Benjamini & Hochberg, Reference Benjamini and Hochberg1995) across the two age algorithms.
Results
Associations between chronological age, DNAm age, and DNAm age residuals
Chronological age was strongly correlated with both Horvath age and GrimAge at each time point (meta rs = 0.86–0.89, meta ps < 0.001; Supplementary Table S1). At the individual cohort level, lower correlations were observed in ArmySTARRS and MRS (Supplementary Table S1), which was likely due to the smaller variance in chronological age in these cohorts (Table 1). Raw Horvath age and GrimAge were also strongly associated with each other at each time point (meta rs = 0.80 and 0.78 at T1 and T2, respectively; Supplementary Table S1). However, Horvath age and GrimAge residuals were weakly correlated with each other at each time point (both meta rs = 0.09; Supplementary Table S1). Meta-analysis revealed a strong correlation between the raw Horvath age and GrimAge estimates with themselves across T1 and T2 (meta rs = 0.91 and 0.96, meta ps < 0.001; Table 2). The residuals were also consistent over time: T1 versus T2 Horvath DNAm age residuals meta-r = 0.65 and GrimAge residuals meta-r = 0.88 (Table 2).
Table 2. Longitudinal correlations (T1 to T2) among DNAm age, DNAm age residuals, and cell type proportions
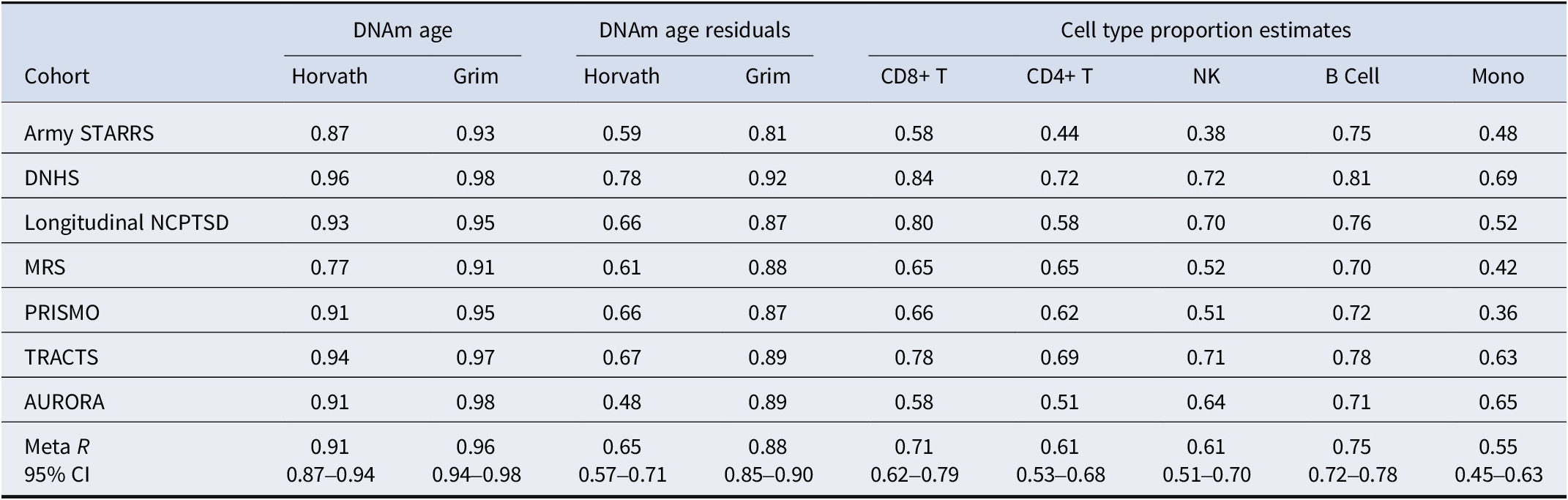
Note: Correlations represent the Pearson correlation coefficients calculated between the same variable at T1 and T2. The 95% confidence intervals were computed for the meta-analytic correlations. All meta-analytic correlations were significantly different from zero at a p-value threshold of 0.001, as determined by a t-test.
Abbreviations: CD8+ T, CD8+ T cell; CD4+ T, CD4+ T cell; NK, natural killer cell; Mono, monocyte; CI, confidence interval; Army STARRS, The Army Study to Assess Risk and Resilience in Servicemembers; DNHS, The Detroit Neighborhood Health Study; NCPTSD, The National Center for PTSD Study; MRS, The Marine Resilience Study; PRISMO, The Prospective Research in Stress-related Military Operations; TRACTS, The Translational Research Center for TBI and Stress Disorders Study; AURORA, The Advancing Understanding of RecOvery afteR traumA Study.
Cell type proportions and their associations with DNAm age residuals
Estimated cell type proportions were strongly related to themselves over time. The estimated proportion of B cells showed the strongest correlation over time (meta r = 0.75, meta p < 0.001), followed by CD8+ T cells (meta r = 0.71, meta p < 0.001), natural killer cells (meta r = 0.61, meta p < 0.001), CD4+ T cells (meta r = 0.61, meta p < 0.001), and monocytes (meta r = 0.55, meta p < 0.001; Table 2). Additionally, the cell type estimates were weakly correlated with both Horvath age residuals and GrimAge residuals at each time point, with meta-correlations ranging from −0.08 to 0.11 for Horvath age residuals and from −0.21 to 0.05 for GrimAge residuals (Supplementary Table S2).
New-onset PTSD diagnosis and change in DNAm age residuals over time
We examined how T1 DNAm age residuals, new-onset PTSD diagnosis (between T1 and T2), and their interaction predicted T2 DNAm age residuals (n = 745). Meta-analysis revealed a significant effect of T1 age residuals on T2 age residuals for both the Horvath algorithm (meta β = 0.63, meta SE = 0.05, meta p = 3.63 × 10−37) and GrimAge algorithm (meta β = 0.85, meta SE = 0.02, meta p < 2.23 × 10−308; Table 3). New-onset PTSD diagnosis was not associated with either T2 Horvath age residuals (meta β = −0.26, meta SE = 0.24, meta p = 0.27) or T2 GrimAge residuals (meta β = 0.22, meta SE = 0.13, meta p = 0.10; Table 3). Meta-analysis also revealed that the T1 Horvath age residuals × new-onset PTSD diagnosis interaction term was significantly associated with T2 Horvath age residuals, after FDR correction for multiple testing across the two algorithms (meta β = 0.16, meta SE = 0.07, meta p = 0.02, p-adj = 0.03, I 2 = 0; Table 3 and Figure 1). This means that the positive association between Horvath age residuals at the two time points was greater among those with new-onset PTSD diagnosis. When additionally covarying for the smoking score, the interaction effect remained significant (meta β = 0.17, meta SE = 0.07, meta p = 0.02) while the smoking score was not associated with T2 Horvath age residuals (meta p = 0.11). This interaction association was not significant for the GrimAge algorithm (meta p = 0.99; Table 3). The association between the interaction term and T2 Horvath DNAm age residuals was not driven by differences in the baseline Horvath age residuals between new-onset PTSD cases versus those who did not develop PTSD (meta p = 0.16; Supplementary Table S3). This implies that those who developed PTSD at T2 did not simply have higher DNAm age residuals at baseline. Full results for each individual cohort can be found in Supplementary Tables S4 and S5. Follow-up analyses revealed that childhood trauma was not related to T2 DNAm age residuals (Supplementary Materials).
Table 3. New-onset PTSD diagnosis as a predictor of T2 DNAm age residuals: main and interactive meta-analytic results
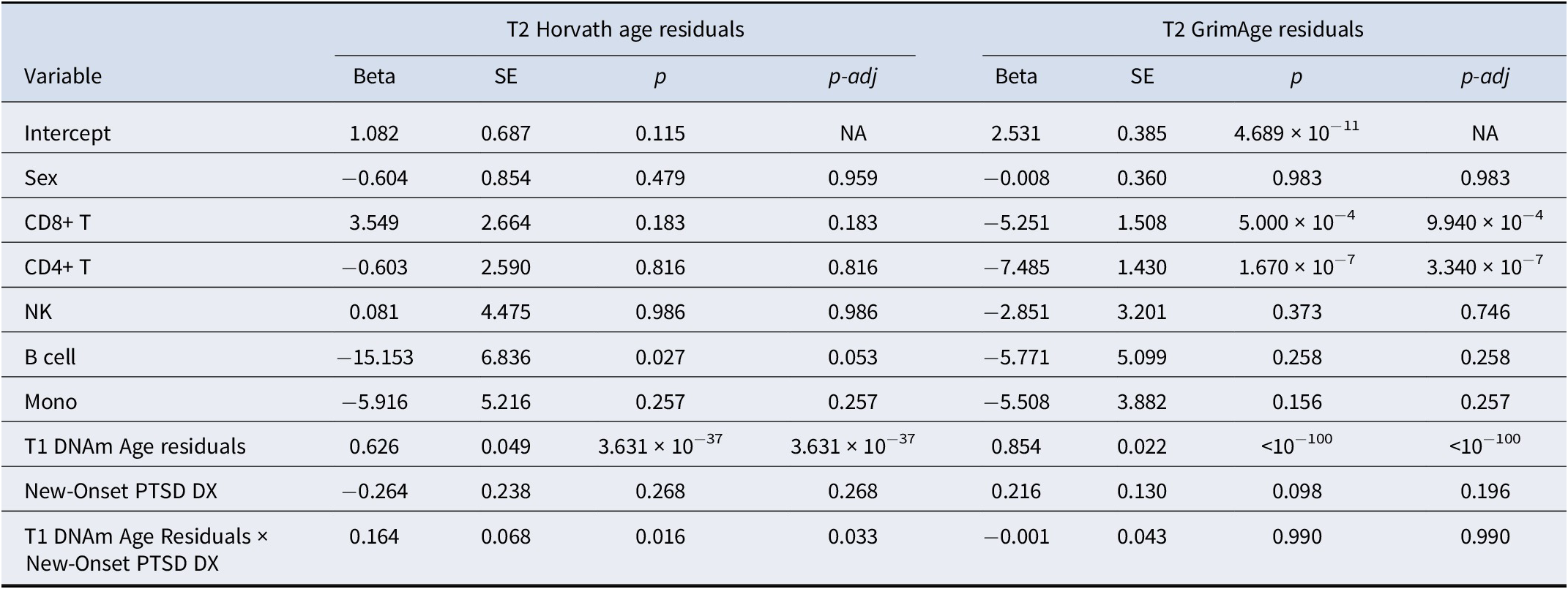
Note: Meta-analytic results from the individual cohort multiple regression linear models. Meta-analytic main effects are derived from the main (and covariate) effect only models in each cohort. Meta-analytic interaction effects are derived from the models with the main and interaction effects. The top three ancestry principal components from each cohort were also included in the model.
Abbreviations: CD8+ T, CD8+ T cell; CD4+ T, CD4+ T cell; NK, natural killer cell; Mono, monocyte; DX, diagnosis; SE, standard error; NA, not applicable; p-adj, p-value adjusted for multiple testing across the two age algorithms using the false discovery rate (FDR) procedure.
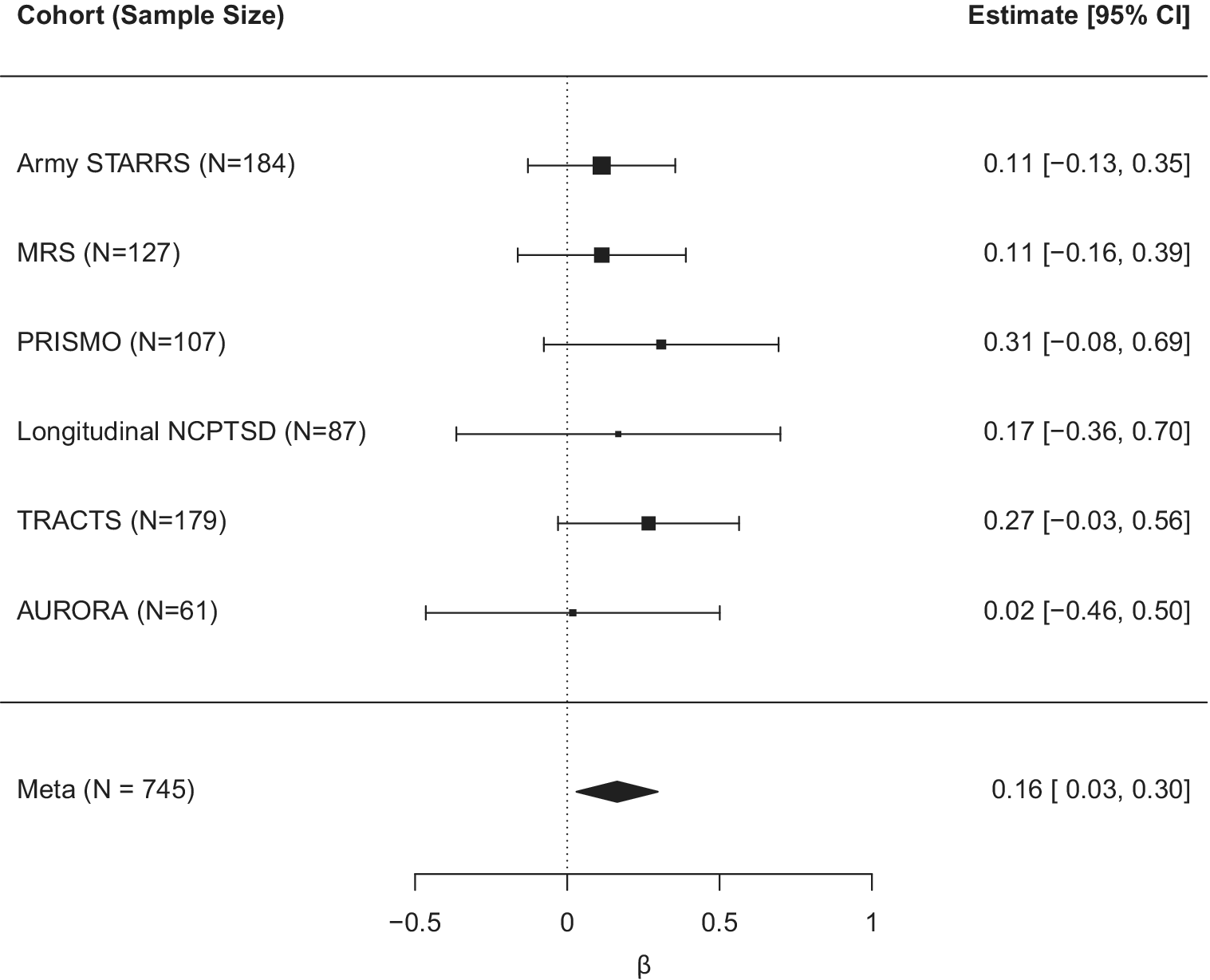
Figure 1. Forest plot for the effect of the interaction term reflecting T1 Horvath DNAm age residuals by new-onset PTSD diagnosis on T2 Horvath DNAm age residuals (controlling for all other main effects in the model).
Change in PTSD symptom severity and change in Horvath DNAm age residuals over time
We next examined if the relationship between Horvath DNAm age residuals over time was moderated by change in PTSD symptom severity over time (n = 1191) as a follow-up to the new-onset PTSD diagnosis analysis. We included the main and interactive effects of baseline Horvath age residuals and change in PTSD symptom severity over time as a predictor of T2 Horvath age residuals (along with covariates). The association between T1 and T2 Horvath age residuals was significant (meta β = 0.63, meta SE = 0.05, meta p = 6.42 × 10−31), but the change in PTSD symptom severity score was not associated with T2 Horvath age residuals (meta β = 0.47, meta SE = 0.63, meta p = 0.46). The interaction between T1 Horvath age residuals and change in PTSD symptom severity over time was significantly associated with T2 Horvath DNAm age residuals (meta β = 0.24, meta SE = 0.12, meta p = 0.05, I 2 = 0; Table 4 and Figure 2). The relationship between DNAm age residuals over time became stronger (a steeper slope) among those with the greatest increase in PTSD symptom severity from T1 to T2. The interaction effect remained significant when further adjusting for the smoking score (meta β = 0.25, meta SE = 0.12, meta p = 0.04), while the smoking score was not associated with T2 Horvath age residuals (meta p = 0.28). The association between the interaction term and T2 Horvath age residuals was not accounted for by baseline differences in T1 Horvath age residuals as a function of change in PTSD symptom severity (meta p = 0.41; Supplementary Table S3). Full results for each individual study are listed in Supplementary Table S6. Although we did not find a significant interaction between GrimAge residuals and new-onset PTSD diagnosis, we conducted the change in PTSD severity analysis for GrimAge residuals for completeness and report the (null) result in the Supplementary Materials.
Table 4. Change (T2–T1) in PTSD severity as a predictor of T2 Horvath DNAm age residuals: main and interactive meta-analytic results
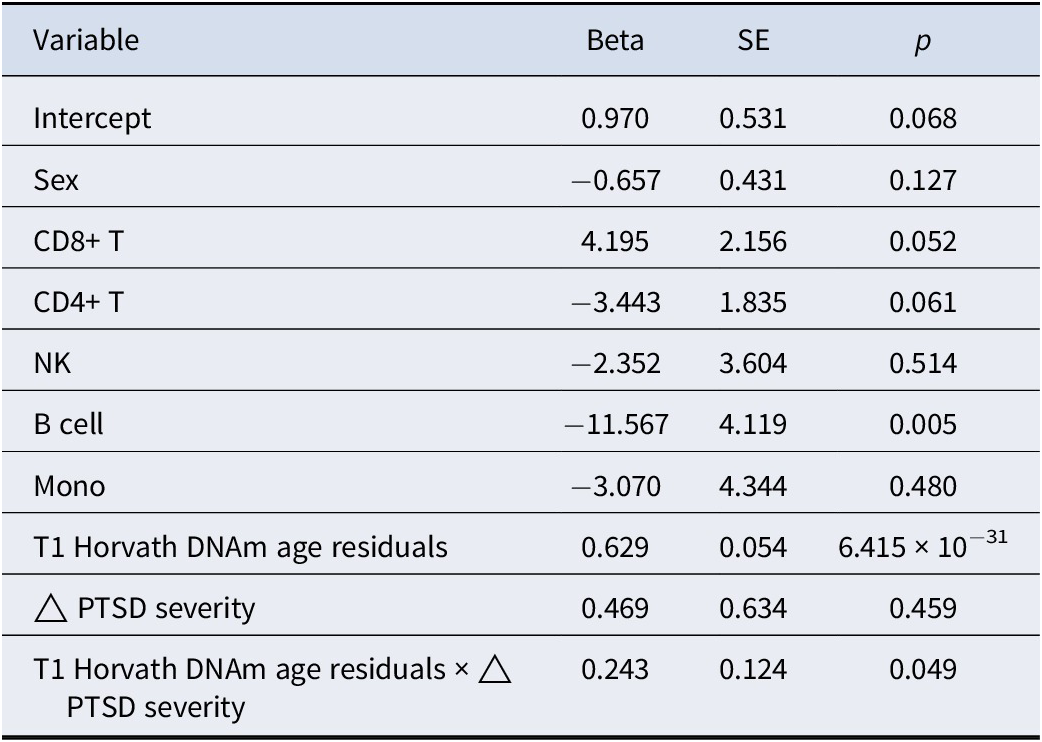
Note: Meta-analytic results from the individual cohort multiple regression linear models. Meta-analytic main effects are derived from the main (and covariate) effect only models in each cohort. Meta-analytic interaction effects are derived from the models with the main and interaction effects. The top 3 ancestry principal components were also included in the meta-analysis. Δ = change (T2–T1).
Abbreviations: CD8+ T, CD8+ T cell; CD4+ T, CD4+ T cell; NK, natural killer cell; Mono, monocyte; SE, standard error.
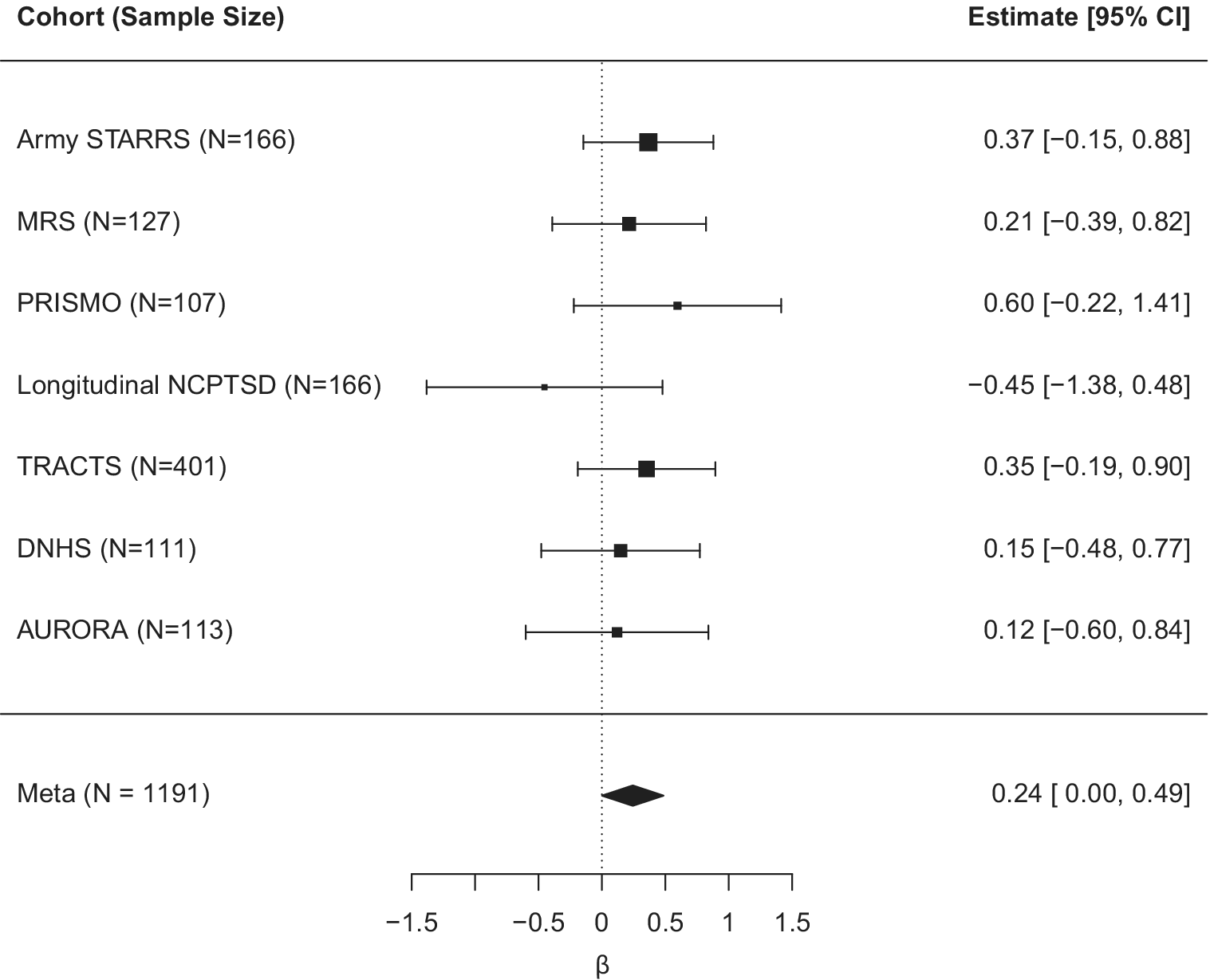
Figure 2. Forest plot for the effect of the interaction term reflecting T1 Horvath DNAm age residuals by change in harmonized PTSD symptom severity on T2 Horvath DNAm age residuals (controlling for all other main effects in the model).
Discussion
The major goals of this study were to first test if individuals who developed new-onset PTSD between two measurements showed greater epigenetic aging than would be expected based on their epigenetic aging at baseline from a time at which they did not meet criteria for PTSD. The second aim was to examine if changes in PTSD symptom severity moderated the strength of the association between epigenetic aging at two time points among individuals with and without PTSD. We expected that individuals with new-onset PTSD and those with increased PTSD symptom severity over time would show higher DNAm age residuals at follow-up than would be expected based on DNAm age residuals at baseline. We evaluated this question via meta-analysis of 7 cohorts. We found that, for every one-year of advanced Horvath DNAm age at baseline, new-onset PTSD cases evidenced an additional 0.16 years of epigenetic aging per the Horvath algorithm at T2 compared to longitudinal controls who did not develop PTSD at T2. In other words, those with new-onset PTSD aged faster by an extra 1.9 months over the time interval, which was on average approximately 18.9 months. Furthermore, an individual experiencing a maximum possible symptom severity change (from having no symptoms at all to having all PTSD symptoms at the most severe level) would have an additional 0.24 years of Horvath age acceleration at T2 compared to an individual with no change in symptom severity score. This implies that this group aged faster by an extra 2.9 months over the time interval, which was on average approximately 21.3 months for the symptom severity analyses.
To further facilitate the interpretation of our findings, we also estimated the pooled standard deviation of the change in harmonized PTSD symptom severity scores across cohorts (SDpooled = 0.23). This allows for the following interpretation of the interaction term: for every one-year increase in Horvath age residuals at baseline, an individual experiencing a pooled SD (23%) increase in PTSD symptom severity at T2 would have an extra 0.68 months of Horvath age acceleration over the average time interval of 21.3 months compared to an individual with no change in symptom severity across the two time points. Although the observed effect sizes were relatively small, the cumulative impact of PTSD on epigenetic aging could become substantial over the course of a lifespan given the inclusion of a large number of young Veterans in their early 30s in this meta-analysis (e.g., ArmySTARRS, MRS, and TRACTS cohorts).
These results are consistent with our hypotheses that age acceleration is increased among individuals with new-onset PTSD and increased symptom severity. No prior studies have examined interactions between new-onset PTSD (or change in symptom severity) by baseline age acceleration, however the structure of these data, with numerous cohorts defined by pre- and post-trauma exposure, made it critical to ask questions regarding how new-onset diagnoses would impact change in metrics of advanced DNAm age. It would not be possible in these cohorts to use baseline PTSD diagnoses to predict change in DNAm age residuals (or the rate of DNAm age change per intervening year) as most individuals were negative for PTSD at the pre-exposure timepoint.
Prior studies have found that PTSD symptom change is associated with changes in DNAm loci that also contribute to Horvath DNAm age and could partially relate to the changes in Horvath DNAm age observed in this study. Specifically, Katrinli et al. (Reference Katrinli, Maihofer, Wani, Pfeiffer, Ketema, Ratanatharathorn and Uddin2022) modeled the association between post-deployment DNAm and change in PTSD symptom severity (PTSS) conditioned on pre-deployment DNAm and found 15 differentially methylated regions (DMRs) associated with change in PTSS. The guanine nucleotide-binding protein, alpha-stimulating activity polypeptide (GNAS) complex locus was one of 15 DMRs associated with PTSD symptom changes and that locus includes a CpG site, cg14597908, which also contributes to the Horvath age calculation (Horvath, Reference Horvath2013). Differential methylation at GNAS has been linked with maternal stress (Vangeel et al., Reference Vangeel, Izzi, Hompes, Vansteelandt, Lambrechts, Freson and Claes2015) and anxiety (Alisch et al., Reference Alisch, Chopra, Fox, Chen, White, Roseboom and Kalin2014, Reference Alisch, Van Hulle, Chopra, Bhattacharyya, Zhang, Davidson and Goldsmith2017), suggesting that it may be sensitive to traumatic stress as well. In a pig model, GNAS was also associated with cellular senescence (Jeon et al., Reference Jeon, Jeong, Kim, Jeong, Hossein, Yang and Hwang2012). Thus, PTSD-related alterations in GNAS DNAm over time could potentially influence the broader changes in advanced epigenetic age observed in this study. Katrinli et al. also reported that PTSS was associated with change in DNAm in genes involved in immune processes and oxidative stress (Katrinli et al., Reference Katrinli, Maihofer, Wani, Pfeiffer, Ketema, Ratanatharathorn and Uddin2022). Alterations in these biological pathways, which also have known associations with accelerated aging (Cevenini, Monti, & Franceschi, Reference Cevenini, Monti and Franceschi2013), could impact the aging process and potentially contribute to the effects observed in this study.
Our primary interaction effects suggested PTSD modulated the slope of age acceleration over time, raising the question of whether PTSD treatment could slow cellular aging and reduce the risk of early onset of age-related disease. However, important challenges exist when attempting to address this question. First, the strongest predictor of follow-up DNAm age acceleration was baseline DNAm age acceleration, so after accounting for this effect, there may be only a small amount of variance remaining to be predicted by treatment. This would suggest that effects of PTSD treatment on cellular aging would be small and sample sizes would need to be large (possibly larger than most intervention trials) to observe an effect. Second, DNAm age estimates may not be sensitive enough to reflect changes in cellular age accurately over the course of a typical 12- or 18-week trial. The study would need to have sufficiently long follow-up periods to observe reliable changes in the DNA methylome. That said, pharmacological studies suggest some promise for effects of treatment on epigenetic aging. For example, a study of 30 individuals with bipolar disorder and 30 healthy controls indicated an association between the use of combination mood stabilizers (lithium carbonate, sodium valproate, and carbamazepine), in contrast to either monotherapy or no medication, with decreased Horvath age acceleration (Okazaki et al., Reference Okazaki, Numata, Otsuka, Horai, Kinoshita, Sora and Hishimoto2020). In human neuroblastoma cells, lithium, valproate, and carbamazepine induced hypermethylation at 377, 70, and 66 CpG sites, respectively, and hypomethylation at 145, 37, and 14 CpG sites, respectively (Asai et al., Reference Asai, Bundo, Sugawara, Sunaga, Ueda, Tanaka and Iwamoto2013). Similar studies are needed in PTSD samples to explore the effect of PTSD treatments and their potential to alter the aging methylome.
Our study also reported high correlations between DNAm age estimates and age residuals with themselves over time, suggesting the robustness of the two age calculators. Both GrimAge and GrimAge residuals were more strongly correlated with themselves over time than were those for the Horvath algorithm. This could be due to its unique 2-step calculation, the inclusion of age and sex in the model (Lu et al., Reference Lu, Quach, Wilson, Reiner, Aviv, Raj and Horvath2019), or that the thousands of GrimAge probes are collectively more stable than the smaller set of probes included in the Horvath model. The stronger correlation between GrimAge residuals over time may be one reason we did not observe a significant interaction between T1 GrimAge residuals and PTSD. There simply may be little variance remaining in the outcome after adjusting for the T1 GrimAge main effect. In addition, the two models may capture different elements of the aging process reflected by methylation and have differential relevance for PTSD-related pathology.
This study also provided the opportunity to test the stability of estimated white blood cell types over time. We found that the cell type proportion estimates were strongly correlated with themselves over time, particularly CD8+ T and B cells. This highlights the robustness of the cell-type estimation algorithm and its value in studying aging-related changes in cell-type composition.
Study limitations
Our results should be considered with several limitations in mind. We did not adjust for intervening time in any analysis because there was little to no variability in time between assessments in the pre- and post-deployment military cohorts. This makes it impossible to determine how correlated measures of epigenetic age are over shorter versus longer periods of time and how factors such as severe, chronic PTSD (i.e., more symptoms over a longer period of time) might alter accelerated aging. Data were only available at two time points so we were unable to test if the pattern of change in epigenetic age is constant or nonlinear over time. Given the absence of baseline PTSD cases in the pre/post deployment samples, it was impossible to explore the association between epigenetic aging and baseline diagnosis and severity.
We did not assess intervening trauma or life stress across the two time points across all cohorts so we could not adjust for new trauma exposure/life stressors or substitute it for new-onset PTSD in our analyses. Thus, we were not able to distinguish the effects of PTSD from trauma or life stress as these variables are often correlated, which is a common challenge. However, a number of prior studies have suggested that PTSD, rather than trauma exposure alone (which is near universal across populations), is more strongly linked to advanced epigenetic age (Wolf et al., Reference Wolf, Logue, Hayes, Sadeh, Schichman, Stone and Miller2016, Reference Wolf, Logue, Morrison, Wilcox, Stone, Schichman and Miller2019; Wolf, Logue et al., Reference Wolf, Logue, Stoop, Schichman, Stone, Sadeh and Miller2018). We suspect that it is the ongoing chronic psychological and physiological stress associated with PTSD (e.g., startle, arousal, anger, and poor sleep) that may have a more direct and sustained influence on physiological processes, including those linked to epigenetic aging.
As information on participants’ medical and pharmacy records were not available, we were unable to identify their associations with epigenetic aging or adjust for them in analyses. We were also unable to adjust for lifestyle factors like body mass index and substance misuse given that these data were not consistently available across cohorts. However, sensitivity analyses revealed that smoking did not account for our primary associations of interest. The error associated with the Horvath age algorithm (3.6-year median absolute difference between the estimated and actual age in the original test data; Horvath, Reference Horvath2013) was greater than the mean time between assessments in some studies (e.g., the AURORA study with a 5.7-month mean interval). It is possible that analyses in cohorts with smaller intervals were more susceptible to measurement error as the algorithm may not be sufficiently sensitive to detect minor changes in methylation over a small period of time. We chose not to analyze the recently developed DunedinPACE metric (Belsky et al., Reference Belsky, Caspi, Corcoran, Sugden, Poulton, Arseneault and Moffitt2022), which uses DNAm data from a single time point to predict pace of aging because we wanted to calculate the observed DNAm age at two timepoints and measure the change in association between them. Due to the limited sample size of civilian participants, we were not able to address whether or how the military versus civilian nature of the samples might influence the findings of this study. While the participants included in this study are diverse in terms of sex, race, ethnicity, military versus civilian status, and geography, they may still not fully represent any particular population, potentially limiting the generalizability of our findings. Finally, although our data are longitudinal, we can make no claims as to a causal association between PTSD and changes in epigenetic age.
Conclusions
This was the first meta-analysis and largest study to date of the associations between PTSD and changes in DNAm age over time. We found meta-analytic evidence across seven cohorts spanning both military and civilian samples that new-onset PTSD diagnosis and increases in PTSD symptom severity were associated with greater age acceleration per the Horvath metric than would be expected based on the baseline measure of age acceleration. This adds to the growing body of evidence suggesting that stress-related disorders may accelerate cellular aging, potentially contributing to the association between traumatic stress and early onset of age-related diseases, such as cardiovascular conditions (Vidal et al., Reference Vidal, Polo, Alvarez, Falgas-Bague, Wang, Lê Cook and Alegría2018) and dementia (Yaffe et al., Reference Yaffe, Vittinghoff, Lindquist, Barnes, Covinsky, Neylan and Marmar2010). This highlights the importance of integrating our understanding of mental and physical health even at the cellular level and underscores the tremendous personal costs associated with traumatic stress.
Supplementary material
The supplementary material for this article can be found at http://doi.org/10.1017/S0033291725000558.
Acknowledgments
PGC-PTSD Epigenetics Workgroup: Allison E Aiello, Allison E Ashley-Koch, Diana Avetyan, Dewleen G Baker, Jean C Beckham, Marco P Boks, Leslie A Brick, Evelyn Bromet, Frances A Champagne, Chia-Yen Chen, Shareefa Dalvie, Nikolaos P Daskalakis, Michelle F Dennis, Segun Fatumo, Catherine Fortier, Sandro Galea, Melanie E Garrett, Elbert Geuze, Gerald Grant, Michael A Hauser, Jasmeet P Hayes, Sian MJ Hemmings, Bertrand Russel Huber, Aarti Jajoo, Stefan Jansen, Seyma Katrinli, Ronald C Kessler, Nathan A Kimbrel, Anthony P King, Joel E Kleinman, Nastassja Koen, Karestan C Koenen, Pei-Fen Kuan, Israel Liberzon, Sarah D Linnstaedt, Adriana Lori, Benjamin J Luft, Jurjen J Luykx, Adam X Maihofer, Christine E Marx, Samuel A McLean, Divya Mehta, William Milberg, Mark W Miller, Janitza Montalvo-Ortiz, Mary S Mufford, Clarisse Musanabaganwa, Jean Mutabaruka, Leon Mutesa, Charles B Nemeroff, Nicole R Nugent, Diana L Núñez-Ríos, Holly K Orcutt, Xue-Jun Qin, Andrew Ratanatharathorn, Sheila A M Rauch, Kerry J Ressler, Victoria B Risbrough, Eugène Rutembesa, Bart P F Rutten, Soraya Seedat, Dan J Stein, Murray B Stein, Sylvanus Toikumo, Robert J Ursano, Annette Uwineza, Leigh L van den Heuvel, Mieke H Verfaellie, Eric Vermetten, Christiaan H Vinkers, Agaz H Wani, Erin B Ware, Derek E Wildman, Erika J Wolf, Ross McD Young, Anthony S Zannas, Xiang Zhao, Ying Zhao, Mark W Logue, Caroline M Nievergelt, Alicia K Smith, Monica Uddin.
Funding statement
This work was supported by R01MH108826 and 2R01MH108826 from NIMH to Smith/Logue/Nievergelt/Uddin. This study was also supported by Merit Review Award Number I01 CX-001276-01 from the US Department of Veterans Affairs Clinical Sciences R&D (CSRD) Service and by the National Institute on Aging of the National Institutes of Health under Award Number RF1AG068121/ 4R01AG068121-02 to Wolf. Katrinli is supported by the BIRCWH under Award Number K12HD085850. This work was also supported by a NARSAD Young Investigator grant (#24135) and a Foundation of Hope for Research and Treatment of Mental Illness grant awarded to Zannas. Funding for the TRACTS study was supported by the Translational Research Center for TBI and Stress Disorders (TRACTS) VA Rehabilitation Research and Development Traumatic Brain Injury National Research Center (B3001-C) to Milberg/Fortier. This work was also supported by the National Institutes for Minority Health and Health Disparities under award 2R01MD011728-01 to Aiello/Uddin/Wildman. Work from Risbrough was supported by BX005872. Work from Rutten was funded by a VIDI award, grant number 91718336 from the Netherlands Scientific Organization. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the US Department of Veterans Affairs or the United States Government.
Disclosure
CYC is an employee of Biogen. SAM served as a consultant for Walter Reed Army Institute for Research, BioXcel, and for Arbor Medical Innovations. NRN serves on the Ilumivu Scientific Advisory Board. KJR has performed scientific consultation for Acer, Bionomics, and Jazz Pharma; serves on Scientific Advisory Boards for Sage, Boehringer Ingelheim, Senseye, Brain and Behavior Research Foundation and the Brain Research Foundation, and he has received sponsored research support from Alto Neuroscience. VBR has performed scientific consultation for Engrail and Jazz Pharmaceuticals and is on the Scientific Advisory Boards for Anxiety and Depression Association of America and Brain Behavior Research Foundation. JG is paid for editorial work for the journal Complex Psychiatry. NPD has served on scientific advisory boards for BioVie Pharma, Circular Genomics and Sentio Solutions for unrelated work. SAMR receives support from the Wounded Warrior Project (WWP), Department of Veterans Affairs (VA), National Institute of Health (NIH), McCormick Foundation, Tonix Pharmaceuticals, Woodruff Foundation, and Department of Defense (DOD). SAMR receives royalties from Oxford University Press and American. DJS has received consultancy honoraria from Discovery Vitality, Johnson & Johnson, Kanna, L’Oreal, Lundbeck, Orion, Sanofi, Servier, Takeda and Vistagen. MBS has in the past 3 years received consulting income from Acadia Pharmaceuticals, Aptinyx, atai Life Sciences, BigHealth, Biogen, Bionomics, BioXcel Therapeutics, Boehringer Ingelheim, Clexio, Delix Therapeutics, Eisai, EmpowerPharm, Engrail Therapeutics, Janssen, Jazz Pharmaceuticals, NeuroTrauma Sciences, PureTech Health, Sage Therapeutics, Sumitomo Pharma, and Roche/Genentech. MBS has stock options in Oxeia Biopharmaceuticals and EpiVario. MBS has been paid for his editorial work on Depression and Anxiety (Editor-in-Chief), Biological Psychiatry (Deputy Editor), and UpToDate (Co-Editor-in-Chief for Psychiatry). MBS has also received research support from NIH, Department of Veterans Affairs, and the Department of Defense. MBS is on the scientific advisory board for the Brain and Behavior Research Foundation and the Anxiety and Depression Association of America. All other authors report no biomedical financial interests or potential conflicts of interest.









 Open access
Open access