

A drug interaction, defined as the modification of the action of one drug by another, can be beneficial or harmful, or it can have no significant effect. An appreciation of clinically important interactions is becoming increasingly necessary with the rising use of combinations of drugs in the management of chronic medical conditions. This trend is likely to increase as the population ages and treatments for a greater number of conditions are introduced into clinical practice. Interactions are a particular problem with elderly people, who, as well as being more likely to take several drugs concurrently, are at greater risk of an adverse drug interaction than younger people. This is because of effects of ageing on organs that deal with the metabolism and excretion of drugs, notably the liver and kidneys. Other vulnerable groups include polydrug misusers, psychiatric patients taking high doses of medication in the management of treatment-resistant disorders and people in developing countries in which there is a high prevalence of self-medication and irresponsible dispensing by a small minority of pharmacists (Reference Edwards and KingEdwards, 2004). Adverse drug interactions can cause significant morbidity and mortality and, as a result of changes in prescribing habits, problems related to polypharmacy are likely to grow.
There are numerous known and potential interactions with psychotropic drugs, and many of them do not have clinically significant consequences. Most clinically important interactions involve drugs that have a narrow therapeutic index (a small difference between the therapeutic and toxic concentrations), for example lithium, phenytoin and warfarin (Table 1 and Box 1). It is impossible to remember every potential interaction, or even those that are clinically significant. However, if the clinician has an understanding of the mechanisms underlying drug interactions, it is more likely that the possible consequences will be considered when selecting therapy.
Table 1 Types and examples of drug interactions

| Interaction type | Example |
|---|---|
| Pharmacodynamic | |
| Direct | Tricyclic antidepressant + monoamine oxidase inhibitor → CNS toxicity |
| Indirect | Selective serotonin reuptake inhibitor + aspirin → increased risk of gastrointestinal bleeding |
| Pharmacokinetic | |
| Absorption | Beneficial: charcoal adsorbs tricyclic antidepressant → decreased absorption of tricyclic overdose → decreased plasma concentration → less toxicity Undesirable: antacids → decreased absorption of phenothiazines → decreased plasma concentration and therapeutic effect of phenothiazines |
| Distribution | Diazepam displaces phenytoin from plasma proteins → increased plasma concentration → increased side-effects of phenytoin |
| Metabolism | Carbamazepine → induction of CYP3A4 → increased metabolism → decreased plasma concentration of risperidone → decreased therapeutic effect of risperidone Protease-inhibiting antiviral drugs → inhibition of CYP3A4 → increased plasma concentration of thioridazine → ventricular arrhythmias |
Box 1 Pharmacological and clinical considerations of psychotropic interactions
Pharmacological
-
• Most interactions are mediated via pharmacokinetic and pharmacodynamic mechanisms
-
• Pharmacokinetic interactions occur when one compound alters the absorption, distribution, metabolism or excretion of another
-
• Induction of liver enzymes by one drug may increase the rate of metabolism and thus decrease the plasma concentration and therapeutic effect of another
-
• Inhibition of liver enzymes by one drug may decrease the rate of metabolism and thus increase the plasma concentration and risk of toxicity of another
-
• Interactions are influenced by many factors, including the dose of the drugs, the genetic make-up, age and diet of the patient, and comorbidity
-
• The effect of inhibition of a particular P450 enzyme is similar to that of administering a drug to someone with a genetic deficiency of that enzyme
-
• Pharmacodynamic interactions occur when two or more drugs that have synergistic or antagonistic actions act on the same receptor or target organ
-
• Potentially hazardous interactions are more likely to occur with drugs that have a narrow therapeutic index and in patients with impaired hepatic or renal function
Clinical
-
• Most interactions are harmless and of only theoretical interest
-
• Some interactions may be beneficial, e.g. in augmentation therapy
-
• The evidence for many potential interactions is based only on in vitro or animal research, single case reports or small-scale uncontrolled studies
-
• Interactions are of particular importance in individuals receiving polypharmacy, in multiple substance misusers and in countries where there is much self-medication and uncontrolled or unethical dispensing
Drug interactions are usually classified as pharmaceutical, pharmacodynamic and pharmacokinetic (Table 1).
Pharmaceutical interactions
Pharmaceutical interactions occur when drugs are mixed outside the body prior to administration. For example, mixing chemically incompatible drugs before intravenous infusion can result in precipitation or inactivation. An example is the incompatibility of phenobarbital with chlorpromazine or opioid analgesics when mixed in the same syringe. Of the three mechanisms, pharmaceutical interactions are least likely to cause problems in clinical practice, and there are no potentially hazardous interactions of this type with psychotropic drugs.
Pharmacodynamic interactions
The most common interactions encountered in clinical practice are pharmacodynamic. They occur when drugs compete for the same receptor or produce antagonistic or synergistic effects on the same target organ or system. Many instances of antagonism are beneficial: for example, naloxone is a specific antagonist that reverses the action of morphine by competing with it for occupancy of the opioid μreceptor. By contrast, antipsychotic drugs reduce the efficacy of levodopa in Parkinson’s disease by blockade of dopamine receptors in the corpus striatum.
Synergistic interactions may be used therapeutically, for example in augmentation treatment of resistant depression with lithium and an anti-depressant, but often they are adverse. A common result is toxicity of the central nervous system (CNS) and hypertension or hypotension, which is discussed in more detail below, in the subsection ‘Effects on the central nervous system’. Depression of the CNS can also occur when alcohol and tricyclic antidepressants are taken concomitantly. Selective serotonin reuptake inhibitors (SSRIs) increase the risk of gastrointestinal bleeding when taken with aspirin or other non-steroidal anti-inflammatory drugs, because of a synergistic inhibition of platelet aggregation.
Pharmacokinetic interactions
Pharmacokinetic interactions occur when the absorption, distribution or elimination of one drug is influenced by another.
Absorption
Important clinical effects caused by changes in drug absorption are rarely seen in general medical or psychiatric practice. These interactions usually result from the binding of two drugs in the gut, preventing their absorption. This property is used therapeutically when activated charcoal is given following an overdose of tricyclic antidepressants. Charcoal adsorbs the drug in the gut, and thereby attenuates the effects of the overdose. An example of an undesirable interaction is the decreased absorption of phenothiazines or sulpiride when they are taken concurrently with antacids, leading to a reduced antipsychotic effect.
One drug may alter the rate of absorption of another. When paracetamol is taken with metoclopramide, the more rapid gastric emptying decreases the time to absorption. Conversely, drugs with antimuscarinic activity, such as tricyclic anti-depressants, delay gastric emptying and thus the rate of absorption of co-prescribed medication. However, for most drugs, other than analgesics, the rate of absorption is not critical to their efficacy.
Distribution
Protein binding
The most frequently recognised mechanism of interactions involving drug distribution is through altered protein binding. Many psychotropic drugs are bound to plasma proteins, but it is the non-protein-bound portion of the drug that is metabolically active. Reduced protein binding increases the free drug fraction and therefore the effect of the drug. Drugs that are highly protein bound (>90%), such as phenytoin, are most prone to interactions mediated by this mechanism. For example, diazepam displaces phenytoin from plasma proteins, resulting in an increased plasma concentration of free phenytoin and an increased risk of adverse effects. The effects of protein displacement are usually not of clinical significance in either general medical or psychiatric practice, as the metabolism of the affected drug increases in parallel with the free drug concentration. The result is that, although the plasma level of the free drug rises briefly, the increased metabolism rapidly restores the level to the previous steady state. Therefore any untoward effects of the interaction are normally short-lived.
Lysosomal trapping
Recently another mechanism of interaction involving drug distribution at a cellular level has been described. Tricyclic anti-depressants, SSRIs and aliphatic phenothiazines are basic lipophilic compounds that are taken up by acidic compartments in the cell. For some drugs, this principally involves association with phospholipids in the cell membrane, whereas others undergo lysosomal trapping within the cell. Tissues such as the lungs, liver and kidneys are rich in lysosomes (intracellular organelles containing lytic enzymes) and if a drug is susceptible to trapping, these tissues take up most of the drug in the body. Drugs that are trapped by lysosomes compete with each other for uptake into the organelles. Mutual inhibition of lysosomal trapping results in higher plasma drug concentrations. This will have the greatest effect on tissues with a low density of lysosomes, such as the heart. Organs with a low concentration of lysosomes would normally be exposed to low concentrations of drugs that undergo lysosomal trapping, but will have increased exposure if drug uptake is limited in tissues with a high concentration of lysosomes. This interaction may contribute to the increased cardiotoxicity of drugs such as thioridazine when co-prescribed with antidepressants (Reference DanielDaniel, 2003).
Within the brain, differences in lysosomal density among the various cells may also predispose to adverse drug interactions. Lysosomes are more numerous in neurons than astrocytes, and decreased trapping may increase exposure of cell surface receptors to the drug. It is not known whether this mechanism contributes to psychotropic drug interactions (Reference DanielDaniel, 2003).
Metabolism
Interactions involving drug metabolism are being increasingly well characterised. Induction of enzymes involved in drug metabolism results in reduced plasma concentrations of drugs that are substrates for the enzyme, and therefore their effectiveness is decreased. For example, enzyme induction by carbamazepine decreases the effectiveness of tricyclic antidepressants and antipsychotics. However, enzyme induction does not usually cause clinically hazardous interactions. Most significant drug interactions involve inhibition of enzyme systems, which increases plasma concentrations of the drugs involved, in turn leading to an increased risk of toxic effects.
The most important enzymes involved in drug interactions are members of the cytochrome P450 (CYP) system that are responsible for many of the phase 1 biotransformations of drugs. These metabolic transformations, such as oxidation, reduction and hydrolysis, produce a molecule that is suitable for conjugation. The potential for interactions involving uridine diphosphate glucurono-syltransferases (UGTs), responsible for phase 2 conjugation reactions, is now recognised. These reactions involve formation of a covalent bond between the drug and an endogenous substrate such as glucuronide, enabling the compound to be eliminated from the body usually by the kidney or in the bile.
Many psychotropic drugs have a high affinity for one or more of the enzymes in the CYP or UGT systems, which play a major role in their metabolism. Induction and inhibition of the activity of drug-metabolising enzymes, and the potential to precipitate hazardous drug interactions, are considered below, in the section ‘Interactions involving drug-metabolising enzymes’.
Excretion
Most clinically significant drug interactions involving excretion relate to the kidneys. The most important of these in psychiatric practice are interactions with lithium. Lithium is filtered by the kidney and reabsorbed by the proximal renal tubule in parallel with sodium. A sustained increase in urinary sodium excretion such as that produced by thiazide diuretics promotes a compensatory reabsorption of sodium by the proximal renal tubule. Lithium reabsorption is similarly enhanced, and because it has a narrow therapeutic index this can increase the plasma lithium concentration to potentially toxic levels.
P-glycoprotein
A further mechanism underlying pharmacokinetic drug interactions has recently been characterised. This involves a specific cell membrane transport protein known as P-glycoprotein (P-gp). The mechanism does not fit neatly into the conventional classification of pharmacokinetic interactions, as P-glycoprotein is involved in drug absorption, distribution and excretion. Drug interactions involving P-glycoprotein are considered in greater detail below.
Interactions involving drug-metabolising enzymes
Many drugs have a lipophilic structure that facilitates their passage through cell membranes to their sites of action. However, lipophilic molecules are less readily excreted from the body and they are usually metabolised to hydrophilic derivatives, which are more easily eliminated.
Cytochrome P450 (CYP) enzymes
A system to standardise the nomenclature of the CYP system was adopted in 1996 (Reference Nelson, Koymans and KamatakiNelson et al, 1996). The three characters that follow the CYP abbreviation (for example in CYP2A6) represent the family, subfamily and individual enzyme, respectively. Families of enzymes share more than 40% homology in their gene sequences (in this example the family is represented by the number 2). Subfamilies share more than 55% homology in their gene sequences (in this example represented by the letter A).
Eleven CYP enzymes are responsible for metabolising the majority of pharmacological agents. Those of importance in the metabolism of psychotropic drugs are CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4, the last being responsible for the metabolism of more than 90% of psychotropic drugs that undergo hepatic biotransformation. Some key features of CYP enzymes are outlined in Box 2.
Box 2 Cytochrome P450 (CYP) enzymes: key points
-
• CYP enzymes are distributed widely among animals and humans and are involved in the oxidation of xenobiotics (foreign substances) and drugs
-
• These enzymes are located in the liver, gut mucosa, lungs, kidney, brain and skin
-
• The enzymes are given a number for the family, a letter for the subfamily and a number for the individual enzyme, e.g. CYP3A4
-
• Those of importance in interactions with psychotropic drugs are CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4
-
• Each enzyme is influenced by genetic, constitutional and environmental factors
-
• Specific drugs have a high affinity for one particular CYP enzyme but most are oxidised by more than one
‘Metabolisers’
Genetic variability plays a major part in the activity of the CYP system and produces differences in the amount or activity of a particular enzyme. Pharmacogenetic polymorphism exists when a variant of a CYP gene generates an enzyme with substantially different activity, and it is present in more than 1% of the population (Reference MeyerMeyer, 1991). Such polymorphism is clinically important as it can result in drug toxicity or ineffectiveness when drugs are given in standard doses. The CYP enzymes that demonstrate pharmacogenetic polymorphism include CYP2C9, CYP2C19 and CYP2D6 (Reference Rogers, Nafziger and BertinoRogers et al, 2002). In clinical practice, the polymorphism produces distinct phenotypes, described as poor metabolisers, extensive metabolisers (the most common type) and ultra-rapid metabolisers.
Poor metabolisers
People who have dysfunctional or inactive CYP enzymes are phenotypically poor metabolisers. The metabolism and elimination of drugs that are substrates for the deficient enzymes are decreased and the likelihood of drug toxicity is increased. By contrast, some compounds are prescribed as pro-drugs (an inactive molecule that is converted to the active substance in the body). Since the therapeutic effect of such a drug depends on its conversion to an active metabolite, in poor metabolisers the drug may be less effective.
Extensive metabolisers
These people are most common in the general population and have normal CYP enzyme activity. They show the predicted responses to therapeutic doses of medication.
Ultra-rapid metabolisers
Ultra-rapid metabolisers have higher than normal activity of CYP enzymes, owing to gene duplication (Reference NortonNorton, 2001). In these individuals, drugs that are substrates for these enzymes may have a markedly reduced or completely absent therapeutic effect. By contrast, the increased metabolism can lead to toxicity when a pro-drug is administered, as it is rapidly transformed into an active metabolite with toxic properties.
CYP enzyme induction and inhibition
CYP enzymes can be induced or inhibited by drugs or other biological substances, with a consequent change in their ability to metabolise drugs that are normally substrates for those enzymes. However, exposure to an enzyme inducer or inhibitor does not always result in altered responses to co-prescribed drugs that are potentially subject to interactions. The probability of a clinically important interaction with co-prescribed drugs is unpredictable in individuals. For example, an enzyme inhibitor is more likely to have a greater effect if the person is an ultra-rapid metaboliser and will have little effect in someone who is already a poor metaboliser. Many drugs are metabolised by several CYP enzymes, so the probability of an interaction will depend on the enzyme(s) affected by the inducer or inhibitor and the availability of an effective alternative route of metabolism in that individual.
Induction
CYP1A2 and CYP3A4 enzymes are capable of being induced, resulting in increased quantities of enzyme as well as increased enzyme activity. Any drug that is a substrate of that enzyme will be more rapidly metabolised and this may result in reduced efficacy. As with ultra-rapid metabolisers, a pro-drug may be activated sufficiently rapidly to produce toxic levels of the active derivative. The onset and offset of enzyme induction take place gradually, usually over 7–10 days, and both the rate and extent can be difficult to predict (Reference Cupp and TracyCupp & Tracy, 1997). The slow onset is due to the time taken for the inducing agent to accumulate and the time needed to synthesise the new enzyme. The slow offset depends on the elimination of the inducing agent and the decay of the increased enzyme levels.
Inducers of the CYP system are less numerous than inhibitors. The most important are inducers of CYP3A4 and include carbamazepine, phenobarbital, phenytoin, rifampicin and St John’s wort (Hypericum perforatum). An example of an interaction in psychiatric practice is the reduced efficacy of haloperidol when carbamazepine is started, resulting from induction of CYP3A4.
Inhibition
Inhibition of CYP enzymes is the most common mechanism that produces serious and potentially life-threatening drug interactions (Reference Johnson, Newkirk and WhiteJohnson et al, 1999). Most enzyme inhibitors act specifically on individual CYP enzymes, so a drug inhibiting CYP2A6 may have no effect on CYP2C19.
Inhibition is usually due to a competitive action at the enzyme’s binding site. Therefore, in contrast to enzyme induction, the onset and offset of inhibition are dependent on the plasma level (and therefore the plasma half-life) of the inhibiting drug. Thus, drugs with a short half-life such as cimetidine will cause rapid inhibition, but the effects will be short-lived after it has been stopped. Important inhibitors of CYP that are involved in psychotropic drug interactions are listed in Table 2. An example of this type of interaction is the increased risk of serious skin reactions with lamotrigine when it is co-prescribed with sodium valproate, resulting from inhibition of CYP3A4.
Table 2 Examples of CYP enzyme inhibitors

| Enzymes inhibited | ||||
|---|---|---|---|---|
| Drug | 1A2 | 2C9, 2C19 | 2D6 | 3A4 |
| Amiodarone | + | + | + | |
| Cimetidine | + | + | + | + |
| Clarithromycin | + | + | ||
| Fluoxetine | + | + | + | |
| Fluvoxamine | + | + | + | + |
| Indinavir | + | |||
| Itraconazole | + | |||
| Ketoconazole | + | + | + | |
| Omeprazole | + | |||
| Ritonavir | + | + | ||
| Sodium valproate | + | |||
Rarely, the inhibition is non-competitive and the inhibiting drug binds irreversibly to the CYP enzyme, forming an inactive complex. This happens, for example, with erythromycin. The offset of inhibition is slow since new enzyme must be synthesised to replace the inactive complexes.
Uridine diphosphate glucuronosyltransferases
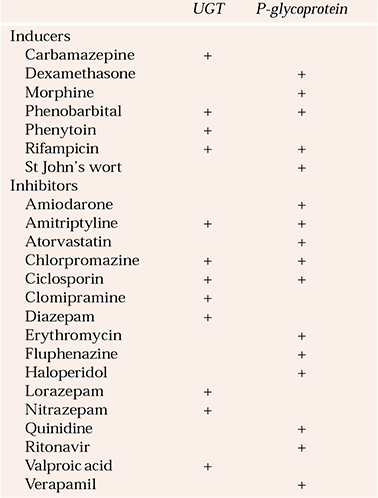
Uridine diphosphate glucuronosyltransferases (UGTs) have received less attention than the CYP enzymes. They are responsible for metabolism of many anxiolytics, antidepressants, mood stabilisers and antipsychotics. Psychotropic inhibitors and inducers of UGTs are shown in Table 3. Inhibition of the metabolism of carbamazepine by valproic acid in part results from an effect on UGTs. Amitriptyline and clomipramine decrease the metabolism of morphine and may contribute to opioid toxicity. The psychopharmacological significance of this type of interaction is not well understood (Reference Kiang, Ensom and ChangKiang et al, 2005).
Table 3 Inducers and inhibitors of uridine diphosphate glucuronosyltransferases (UGTs) and P-glycoprotein
| UGT | P-glycoprotein | |
|---|---|---|
| Inducers | ||
| Carbamazepine | + | |
| Dexamethasone | + | |
| Morphine | + | |
| Phenobarbital | + | + |
| Phenytoin | + | |
| Rifampicin | + | + |
| St John’s wort | + | |
| Inhibitors | ||
| Amiodarone | + | |
| Amitriptyline | + | + |
| Atorvastatin | + | |
| Chlorpromazine | + | + |
| Ciclosporin | + | + |
| Clomipramine | + | |
| Diazepam | + | |
| Erythromycin | + | |
| Fluphenazine | + | |
| Haloperidol | + | |
| Lorazepam | + | |
| Nitrazepam | + | |
| Quinidine | + | |
| Ritonavir | + | |
| Valproic acid | + | |
| Verapamil | + | |
P-glycoprotein
P-glycoprotein is a member of the adenosine triphosphate (ATP) binding cassette (ABC) transporter superfamily. It is an efflux pump capable of transporting a wide range of compounds from the intracellular space into the extracellular matrix. It is present in tissues throughout the human body, such as the intestinal wall, liver, kidneys and cerebral microvascular endothelium, and it plays a significant role in drug absorption, distribution and elimination.
Intestinal P-glycoprotein reduces effective drug absorption by actively transporting drugs back into the intestinal lumen. P-glycoprotein in the liver and kidneys promotes excretion of drugs from the blood stream into the bile and urine, respectively. In addition, P-glycoprotein is present at the blood–brain barrier, where it reduces drug access to the CNS.
Like CYP enzymes, P-glycoprotein can be induced and inhibited by other drugs (Table 3), which creates the potential for drug interactions (Reference KimKim, 2002). This is best illustrated by loperamide, an opioid derivative that normally does not cause central effects, owing to its exclusion at the blood–brain barrier. However, if it is co-administered with quinidine, an inhibitor of P-glycoprotein, it causes CNS side-effects. In animal models, the CNS concentrations of certain drugs increase by 10- to 100-fold when P-glycoprotein is inhibited (Reference Lin and YamazakiLin & Yamazaki, 2003). The role of P-glycoprotein in mediating drug interactions with psychotropic drugs is only starting to be unravelled (Reference Carson, Ousmanou and HoylerCarson et al, 2002).
Serious clinical consequences of drug interactions
The potentially hazardous and life-threatening consequences of the types of interaction with psychotropic drugs that we have discussed are presented below (see also Box 1).
Effects on the CNS
Profound oversedation
Severe sedation due to the additive effect (summation) of drugs with sedating properties is a particular problem in elderly and frail people, and it can lead to falls and injuries (especially fractures of the femoral neck). Excessively drowsy patients are also at increased risk of venous thromboembolism and, if confined to bed, of hypostatic pneumonia. In people who drive, increased sedation due to drug interactions carries a correspondingly increased risk of road traffic accidents. It is the responsibility of the individual not to drive if their ability to do so safely is impaired by drugs, whether prescribed or not. Driving when reactions are impaired by drugs may lead to prosecution.
Profound and prolonged sedation can be brought about by inhibition of CYP3A4 enzymes that are involved in the metabolism of anxiolytics and sedatives. This occurs when one of the protease-inhibiting antiviral compounds amprenavir, indinavir and ritonavir are co-administered with any of the following: alprazolam, clorazepate, diazepam, flurazepam and zolpidem. In each of these cases, the inhibition of metabolism causes high plasma levels of the anti-anxiety and hypnotic substances.
Serotonin syndrome and related CNS toxicity
Central nervous system toxicity refers to a wide range of drug-induced toxic effects, including excitation and restlessness; tremor, rigidity and myoclonus; pyrexia with sweating and flushing; fluctuating vital signs; and delirium. These can progress to stupor, coma and, at worst, death.
The reactions include so-called serotonin syndrome, in which the toxicity is thought to be due to an increased effect of biogenic amines, particularly serotonin (5-hydroxytryptamine, 5-HT). Toxicity can also result from the combined use of drugs that increase monoamine neurotransmission. Examples are the toxicity resulting from the concomitant prescribing of monoamine oxidase inhibitors (MAOIs) with SSRIs, tricyclic and related anti-depressants, tryptophan or St John’s wort. It can also occur when any of these drugs is co-prescribed with:
-
• clozapine
-
• the oxazolidinone antibacterial linezolid (which is a reversible non-selective MAOI)
-
• the appetite suppressant sibutramine (which inhibits the reuptake of noradrenaline and serotonin)
-
• agonists (rizatriptan, the anti-migraine 5-HT1 sumatriptan and zolmitriptan)
-
• tetrabenazine (whose action in patients with movement disorders may be due to dopamine and serotonin depletion at nerve endings)
-
• entacapone and selegiline (which inhibit monoamine breakdown)
-
• the analgesics dextromethorphan and pethidine (the mechanism is uncertain).
Convulsive seizures
Seizures may result from the additive effects of two or more drugs that lower the convulsive threshold, as occurs for example when fluvoxamine or maprotiline are prescribed for patients taking clozapine. They may also result from inhibition of metabolism of a drug with epileptogenic properties. For instance, when erythromycin is prescribed for someone receiving clozapine, the antibiotic inhibits CYP3A4, thereby decreasing the metabolism and increasing the plasma concentration of the antipsychotic. This increases the risk of seizures. Alternatively, seizures may occur in people with epilepsy as a result of decreased plasma concentration of an anti-epileptic owing to enzyme induction. For instance, St John’s wort can increase the metabolism, and thus decrease the plasma concentration, of carbamazepine and phenytoin, resulting in inadequate control of the epilepsy.
Effects on the cardiovascular system
Hypotension
Although it is often considered a minor unwanted effect of psychotropic drugs, hypotension can be hazardous, especially in elderly people. As with oversedation, the drop in blood pressure can cause falls and injuries. Rarely, it can lead to cerebral ischaemia and stroke, an organic confusional state or myocardial ischaemia (which can precipitate a myocardial infarct).
Hypotension is a well-recognised anti-adrenergic effect of many pharmacotherapeutic agents. It is more liable to occur (as a result of summation) with combinations of drugs that have hypotensive effects, as when MAOIs are co-prescribed with drugs such as
-
• antihypertensive agents
-
• pethidine
-
• the non-opioid analgesic nefopam
-
• selegiline.
In the case of nefopam, the fall in blood pressure is possibly greater because inhibition of its metabolism leads to increased plasma levels of the analgesic.
Hypertension
Hypertension is a serious unwanted effect of psychotropic medication. It is best known in relation to MAOIs – notably in the ‘cheese reaction’. Inhibition of monoamine oxidase in the intestinal tract and liver results in increased plasma levels of amines (especially tyramine) that are derived from certain foods. The MAOIs also inhibit presynaptic mitochondrial monoamine oxidase (which is their therapeutic mode of action), with a consequent increase in the concentration of noradrenaline in the presynaptic vesicles and synaptic cleft. As a result of these actions, MAOIs taken in conjunction with the following drugs may result in hypertension:
-
• tricyclic and related antidepressants (in which case a rise or fall in blood pressure may occur)
-
• oxypertine
-
• buspirone
-
• bupropion (amfebutamone)
-
• the dopaminergics entacapone, levodopa and selegiline.
The reaction has also been reported as a result of the co-prescribing of SSRIs and selegiline (which is an MAO-B inhibitor). The most serious consequences of hypertensive crises are intracerebral bleeding, subarachnoid haemorrhage, coma and death.
Ventricular arrhythmias
Of the various cardiac effects of psychotropic drugs, arrhythmias are the most important. Co-prescribing more than one drug that lengthens the QT interval on the electrocardiogram (ECG) is potentially dangerous. The same risk exists when a drug that increases the QT interval is co-administered with a compound that inhibits its metabolism. The risk is higher in people who have the risk factors shown in Box 3. Prolongation of the QT interval increases the period of vulnerability of the myocardium during which ventricular arrhythmias – particularly the irregular, broad, complex ventricular tachycardia known as torsade de pointes (‘twisting of the points’) – may be precipitated by ventricular premature beats.
Box 3 Risk factors for prolonged QT interval
-
• High doses of an antipsychotic and/or anti-depressant known to affect K+ channels
-
• Treatment with droperidol, pimozide, sertin-dole or thioridazine
-
• Co-administered drugs that prolong the QT interval
-
• Heart disease:
-
• myocardial ischaemia
-
• left ventricular dysfunction or hypertrophy
-
• previous torsade de pointes
-
• ECG abnormalities, including bradycardia, ventricular extrasystole and heart block
-
-
• Hepatic or renal failure
-
• Alcoholic liver disease
-
• Low plasma K+,Ca2+ or Mg2+
-
• Advanced age
-
• Female gender
Prolongation of the QT interval is determined by several factors, especially blockade of the rapid component of the delayed rectifier potassium current (I Kr) responsible for repolarisation of cardiac Purkinje cells and myocardial cells in the later phase of the cardiac action potential (Reference O'Brien and OyebodeO’Brien & Oyebode, 2003). Many drugs, including certain antipsychotics and antidepressants, bind to this potassium channel and thereby decrease the outward movement of potassium, which is responsible for ventricular repolarisation. Some antipsychotics –especially droperidol, pimozide, sertindole and thioridazine – have a greater capacity than others to cause I Kr blockade (Reference Glassman and BiggerGlassman & Bigger, 2001; Reference TaylorTaylor, 2003). Some of these drugs also block other ion channels, thus adding to the complexity of their electrophysiological effects. If these compounds are prescribed for people who already have prolonged repolarisation, such as is produced by many anti-arrhythmic drugs, they increase the risk of ventricular arrhythmias.
Other serious effects
Interactions can increase the risk of serious side-effects of co-administered substances. Examples are an increased risk of:
-
• agranulocytosis with clozapine when its plasma concentration is increased by the co-administration of drugs that inhibit CYP enzymes, for example ritonavir, various antidepressants or cimetidine
-
• lithium toxicity due to an increased plasma lithium concentration brought about by the decreased excretion of the drug that occurs when potassium-sparing diuretics (such as amiloride or spironolactone) and thiazide diuretics, angiotensin converting enzyme (ACE) inhibitors and non-steroidal anti-inflammatory drugs are co-prescribed.
A decreased therapeutic effect caused by drug interactions can occur in various areas of drug treatment. St John’s wort can lead to reduced plasma concentrations of various antibacterial, antiviral, anticoagulant and immunosuppressive drugs, and theophylline, which in turn can decrease their efficacy.
Conclusions
Much research has been carried out into potentially hazardous interactions with psychotropic drugs, yet there is much that remains unknown. The evidence for some potential interactions is based on animal experiments, isolated case reports in which there is doubt about the cause-and-effect relationships, or small-scale volunteer studies that may not reflect the action of, and interaction between, drugs in patients. Other possible interactions are deduced from the occurrence of interactions during treatment with related compounds. There is also a lack of knowledge of the size of the problem and of the many pharmacological and host factors that determine whether or not an individual will have a particular interaction. Despite these gaps in our knowledge, there is now sufficient understanding to help prevent a number of serious and potentially fatal interactions. Particularly important is an awareness of the role of modulation of the activity of CYP enzymes in mediating drug interactions.
Preventive measures include avoidance of unnecessary or unthoughtful polypharmacy, an understanding of the predictors of severe interactions (for example, older age, impaired hepatic and renal function, and multiple substance misuse), better education of prescribers and pharmacists about known and potential interactions, and improved drug safety monitoring directed towards the discovery of new interactions.
MCQs
-
1 A hazardous drug interaction is more likely to occur:
-
a if the drug in question has a wide therapeutic index
-
b with inhibition rather than induction of drug-metabolising enzymes
-
c with drugs that are renally excreted
-
d during long-term therapy with both drugs
-
e in elderly people.
-
-
2 Inhibitors of the cytochrome P450 enzyme system:
-
a affect all enzymes equally
-
b have a slow onset of action on the enzymes
-
c produce predictable effects on the metabolism of drugs that are metabolised by CYP enzymes
-
d include cimetidine and omeprazole
-
e are less likely to produce drug interactions than are inhibitors of UGTs.
-
-
3 P-glycoprotein:
-
a is a hepatic drug-metabolising enzyme
-
b can be inhibited or induced by drugs
-
c is involved in maintenance of the blood–brain barrier
-
d is involved in the renal excretion of lithium
-
e has a well-defined role in the genesis of interactions involving psychotropic drugs.
-
-
4 Elderly mentally ill people who have concomitant physical illnesses:
-
a can be treated safely with simvastatin while taking paroxetine
-
b need pose no concerns if treated with nefopam and an MAOI
-
c are at increased risk of hip fractures while receiving thioridazine and lorazepam
-
d are at greater risk of death if they are taking St John’s wort and require theophylline for a severe asthmatic attack
-
e have an increased chance of developing agranulocytosis if fluvoxamine is co-administered with clozapine.
-
-
5 Patients in general can be:
-
a safely treated with a combination of penicillin and antidepressants that have epileptogenic potential
-
b legally prevented from driving if they are prescribed carbamazepine together with trifluoperazine
-
c in danger of torsade de pointes if they have alcohol dependence and are treated with sertraline
-
d safely co-prescribed lithium and thiazide diuretics
-
e at high risk of hypotension when they are receiving both moclobemide and selegiline.
-
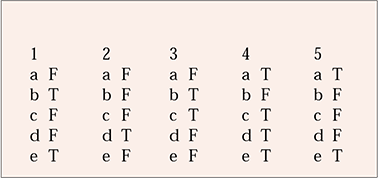
MCQ answers
| 1 | 2 | 3 | 4 | 5 | |||||
|---|---|---|---|---|---|---|---|---|---|
| a | F | a | F | a | F | a | T | a | T |
| b | T | b | F | b | T | b | F | b | F |
| c | F | c | F | c | T | c | T | c | F |
| d | F | d | T | d | F | d | T | d | F |
| e | T | e | F | e | F | e | T | e | T |




eLetters
No eLetters have been published for this article.