


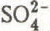 ,
,  ) were of approximately the same magnitude as the daily changes in gains recorded in the snow cover during the melt period. In contrast, the mean value for the contribution by wet deposition to the total loads of K
) were of approximately the same magnitude as the daily changes in gains recorded in the snow cover during the melt period. In contrast, the mean value for the contribution by wet deposition to the total loads of K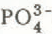 in the snow cover was far outweighed by the gains which were observed at the same time. The expected losses for the snowpack, calculated from the sum of the total loads stocked in the pack at the beginning of the melt period and the total loads in precipitation during the melt period, were lower than the sum of the actual losses observed for all ionic species except H
in the snow cover was far outweighed by the gains which were observed at the same time. The expected losses for the snowpack, calculated from the sum of the total loads stocked in the pack at the beginning of the melt period and the total loads in precipitation during the melt period, were lower than the sum of the actual losses observed for all ionic species except H , Mg
, MgIntroduction
The acidity of atmospheric precipitation in north-eastern North America (Reference Likens and ButlerLikens and Butler 1981) has been identified as the prevailing factor in the acidification of poorly buffered surface waters in both the north-eastern United States and eastern Canada (Reference WrightWright 1983). In the latter region, snow, which covers the ground for 5 to 9 months of the year, accumulates acidic aerosols by both wet and dry deposition during the winter period. The subsequent spring season is characterized by the rapid release of pollutant loads by the snowpack (Reference Johannessen and HenriksenJohannessen and Henriksen 1978) leading to high acidity values in surface waters and concomitant stress conditions for the aquatic biota (Reference Driscoll, Baker, Bisogni and SchofieldDriscoll and others 1980).
Although the major source of the acidic input to the snow cover is the deposition of ![]() and
and ![]() aerosols transported over long distances from the industrial areas of both eastern Canada and eastern and mid-western United States (Reference Altshuller and McBeanAltshuller and McBean unpublished) local characteristics of watersheds also influence the acidity of the snow cover in winter (Reference Jeffries and SnyderJeffries and Snyder 1981, Reference Jones, Sochanska, Bougie and CharetteJones and others 1984). In the Laflamme watershed of Quebec, where conifer stands are predominant, Reference Jones, Sochanska, Bougie and CharetteJones and others (1984) found that, although the concentrations of ionic species (K+ excepted) in incident snowfall was, in general, little affected by the canopy, the concentration of some ionic species (H+, K+,
aerosols transported over long distances from the industrial areas of both eastern Canada and eastern and mid-western United States (Reference Altshuller and McBeanAltshuller and McBean unpublished) local characteristics of watersheds also influence the acidity of the snow cover in winter (Reference Jeffries and SnyderJeffries and Snyder 1981, Reference Jones, Sochanska, Bougie and CharetteJones and others 1984). In the Laflamme watershed of Quebec, where conifer stands are predominant, Reference Jones, Sochanska, Bougie and CharetteJones and others (1984) found that, although the concentrations of ionic species (K+ excepted) in incident snowfall was, in general, little affected by the canopy, the concentration of some ionic species (H+, K+, ![]() ,
, ![]() ) in the snow on the ground showed unexpected temporal and spatial variations. It was suggested that organic debris deposited from the canopy during and between individual precipitation events could be the source of some of the changes observed.
) in the snow on the ground showed unexpected temporal and spatial variations. It was suggested that organic debris deposited from the canopy during and between individual precipitation events could be the source of some of the changes observed.
Dry deposition of micron and submicron acidic aerosols (Reference Ibrahim, Barrie and FanakiIbrahim and others 1983) and larger dust particulates of a local nature (Reference Popp, Jensen, Brandvold, Brandvold and KeithPopp and others 1982) are already known to play a role in the evolution of the chemical composition of snow cover. The local deposition of debris from the canopy, however, can be appreciable in the boreal forest; its presence may dominate other deposition to such an extent that problems are encountered both in sampling procedures and in the subsequent interpretation of the data relating the quality of incident precipitation with that of the snowpack (Reference Jeffries and SnyderJeffries and Snyder 1981, Reference Jones, Sochanska, Bougie and CharetteJones and others 1984).
In the study of the role that snow cover at Lake Laflamme plays in both the retention and the release of acidic pollutants into the stream tributaries at springtime, the influence of the local deposition of organic matter on the snow surface cannot be ignored. Thus, during the spring of 1983 we carried out a snow-cover survey at Lake Laflamme, Quebec, with the purpose of determining the impact of the coniferous stands on the chemistry of the snowpack during both premelt and melt periods. The main objective of this study was to establish the relative importance of wet and dry deposition (atmospheric aerosols, dust and Organic debris) to the total loads of ionic species in the snow cover of boreal forest sites receiving acid precipitation.
Methodology
Study area and sampling sites
The watershed of Lake Laflamme (0.68 km2) lies between 777 (mean lakewater level) and 884 m a.s.1. altitude at lat 47°19', long 71°07' in the Montmorency Forest (mixed spruce, fir and birch) 80 km to the north of the city of Quebec, Canada (Fig.1). The mean annual precipitation is 1400 mm of which 34% is snow. The sampling sites for the study of the snow cover (21 baseline stations and one main station) were located within an area of 2880 m2 (45 × 64 m). Detailed biophysical characteristics of each site are described in Reference Jones, Sochanska, Bougie and CharetteJones and others (1984).

Fig.1. Study site and annual precipitation, Lake Laflamme, Québec.
Precipitation (snow and rain) was collected on an event basis by means of two Sangamo Type A wet-only collectors placed outside the drip zone of conifers. Samples for the chemical analysis of the snow cover were obtained by coring with a clear plastic tube (Plexiglas 2 m long and 7.5 cm internal diameter) (Reference Jeffries and SnyderJeffries and Snyder 1981). No special precautions to remove surface debris from cores were taken except where twigs, lichen clumps and animal excrement interfered with the sampling operations. All samples were placed in plastic bags (polyethylene) and kept at −20°C until melted for analysis. Sampling of the snow cover was started on 15 March 1983 and terminated on 12 May when approximately 40% of the ground cover was bare and from 9 to 10 cm of very dense snow cover remained. The snow cover had completely disappeared by 15 May.
The melt period commenced on 8 April when a large lysimeter (20 m2) collected small discharges, although small lysimeters (0.1, 0.125 m2) showed only appreciable discharges beginning on 25 April (Reference JonesJones 1985). Date from the large lysimeter could not be used to verify directly the discharge of total ionic loads from the snow cover as the lysimeter was subjected to heavy infiltration by surface water during the 1983 melt season.
Analytical procedures
Snow samples were melted under an atmosphere of N2 in large capped plastic bottles. The volumes of the melted samples were recorded and the values used to determine the water equivalent [WE] of each original snow-cover sample. Conductivity and pH were determined on the unfiltered liquid samples by means of a conductivity meter (Radiometer CDM) and a pH meter (Radiometer PHM26), respectively. The samples were subsequently filtered through polycarbonate (47 mm, 0.4 μm) and analyzed for anions (Cl−, ![]() ,
, ![]() ,
, ![]() ) by ion chromatography (Dionex 12S) within 48 h. Cations were conserved in the filtered samples by the addition of nitric acid (0.5 ml 1−1) and
) by ion chromatography (Dionex 12S) within 48 h. Cations were conserved in the filtered samples by the addition of nitric acid (0.5 ml 1−1) and ![]() by the addition of concentrated sulfuric acid (0.2 ml 1−1). Ca2+, Mg2+, Na+, K+. Al and Mn were analyzed by means of atomic absorption spectrophotometers (Ca2+, Mg2+, K+, Na+ by Varian 575 and Al and Mn by Varian G7A95 graphite furnace and Varian 1275) and
by the addition of concentrated sulfuric acid (0.2 ml 1−1). Ca2+, Mg2+, Na+, K+. Al and Mn were analyzed by means of atomic absorption spectrophotometers (Ca2+, Mg2+, K+, Na+ by Varian 575 and Al and Mn by Varian G7A95 graphite furnace and Varian 1275) and ![]() by colorimetry (Technicon AutoAnalyzer II, procedure 154–71W, indophenol-sodium nitroprusside).
by colorimetry (Technicon AutoAnalyzer II, procedure 154–71W, indophenol-sodium nitroprusside).
Detection limits are Cl−: 1 μeq 1−1, ![]() : 1 μeq 1−1,
: 1 μeq 1−1, ![]() : 1 μeq 1−1,
: 1 μeq 1−1, ![]() : 5 μeq 1−1, Ca2+ : 0.5 μeq 1−1, Mg2+ : 0.8 μeq 1−1, Na+ : 0.5 μeq; 1−1, K+ : 0.3 μeq 1−1,
: 5 μeq 1−1, Ca2+ : 0.5 μeq 1−1, Mg2+ : 0.8 μeq 1−1, Na+ : 0.5 μeq; 1−1, K+ : 0.3 μeq 1−1, ![]() : 1 μeq; 1−1, Al μg 1−1 and Mn: 1 μg 1−1.
: 1 μeq; 1−1, Al μg 1−1 and Mn: 1 μg 1−1.
Data analysis
Anionic deficiencies for all samples were calculated from the expression ∑(Cl− + ![]() +
+ ![]() +
+ ![]() +
+ ![]() ) − ∑(H+ +
) − ∑(H+ + ![]() + Ca2+ + Mg2+ + Na+ + K+ + Al3+ + Mn2+) μeq 1−1. At the pH of the samples collected,
+ Ca2+ + Mg2+ + Na+ + K+ + Al3+ + Mn2+) μeq 1−1. At the pH of the samples collected, ![]() concentrations are negligible. Al3+ and Mn2+ were calculated from total soluble Al, total soluble Mn and H+ concentrations according to the method used by PGC Campbell (personal communication).
concentrations are negligible. Al3+ and Mn2+ were calculated from total soluble Al, total soluble Mn and H+ concentrations according to the method used by PGC Campbell (personal communication).
Total ionic loads from wet deposition were calculated as an average value for the two Sangamo collectors on an event and/or daily basis. In the case where a precipitation event was continuous over a period of more than 24 h, the daily loading was calculated by a repartition of the total load deposited according to the percentage volume of the total precipitation that fell in every 24 h period of the event.
The mean value for the total ionic loads (Ix, meq m−2) of the snow cover was calculated from the expression

where [WE] represents the water equivalent (m), [CX] the concentration of the ionic species X (µeq 1−1), and n the total number of cores for baseline stations. For the premelt period, the number of cores taken at any one sampling date varied from 14 to 18. At the beginning of the melt period, seven cores were taken on every sampling date at intervals of two to four days while at the end of the melt period, five cores were taken at any one time every two days. Gains or losses in the total toad of the snowpack during any particular period of time were averaged out over the period as daily rate changes (meq m−2 d−1).
Regrouping of chemical parameters by factor analysis was carried out after axes rotation with Kaiser normalization (Reference Nie, Hull, Jenkins, Steinbrenner and BentNie and others 1975).
Results and Discussion
During the study period, the evolution of the snow cover was divided into five main phases (Fig.2). The first two phases were separated by the first meltwaters issuing from the pack on 8 April 1983. Each of the successive phases was separated from the preceding phase by the rains that occurred on 17 April, 24–25 April and 1–4 May respectively.

Fig.2. Mean water equivalent of the snow cover at Lake Laflamme, spring 1983. *This value (12.2 cm) is overestimated by 12%.
The first phase (20 March to 8 April) was characterized by wet deposition in the form of snowfall (76.8 cm), a very small amount of rain (0.6 mm) in mixed precipitation, low temperatures and a stable pack (accretion stage with no water loss). From a maximum value of 21.2 cm for the water equivalent on 8 April, the net loss of water from the pack due to meltwater discharge up to the rain event of 17 April (second phase) was calculated from the measured water equivalent of the snow cover to be 0.7 cm d−1.
Subsequent to the rain of 17 April, the snowpack entered into the third melt phase in which losses in the total water equivalent were compensated by an input of mixed precipitation (snow and rain) which slightly increased the overall water equivalent of the pack. During the fourth phase (25–30 April), pack decline was 0.5 cm d−1 The fifth phase of pack evolution was characterized by exceptionally heavy rain events (1–4 May and 7–11 May) and net pack discharges increased to 1 cm d−1.
Over the whole period represented by the premelt and melt phases, the mean (arithmetic, non-volume-weighted) ionic concentrations of ![]() ,
, ![]() and H+ in the snow cover were comparable (i.e. snow cores, n = 168;
and H+ in the snow cover were comparable (i.e. snow cores, n = 168; ![]() : 21.6 µeq 1−1 (range 12.7 to 34.5 μeq ℓ−1);
: 21.6 µeq 1−1 (range 12.7 to 34.5 μeq ℓ−1); ![]() : 21.4 µeq 1−1 (range 5.7 to 46.2 μeq 1−1) and H+: 29.5 μeq 1−1 (range 10.0 to 53.1 µeq 1−1)) while the mean concentrations of K+ and
: 21.4 µeq 1−1 (range 5.7 to 46.2 μeq 1−1) and H+: 29.5 μeq 1−1 (range 10.0 to 53.1 µeq 1−1)) while the mean concentrations of K+ and ![]() were lower but fluctuated greatly around the mean values (K+ : 10.8 μeq 1−1 (range below the detection limit (BDL) to 82 µeq 1−1) and
were lower but fluctuated greatly around the mean values (K+ : 10.8 μeq 1−1 (range below the detection limit (BDL) to 82 µeq 1−1) and ![]() : 5 μeq 1−1 (range BDL to 42.1 μeq 1−1)), Large fluctuations in the concentration of these latter ions were particularly evident during rain episodes when K+ and
: 5 μeq 1−1 (range BDL to 42.1 μeq 1−1)), Large fluctuations in the concentration of these latter ions were particularly evident during rain episodes when K+ and ![]() distribution in the pack was heterogeneous.
distribution in the pack was heterogeneous.
The concomitant evolution of gains and losses in total ionic loads of the snow cover and the wet deposition loadings to the snow cover during the main phases of the spring melt are shown in Figures 3 (H+, ![]() ) and 4 (K+,
) and 4 (K+, ![]() ).
).

Fig.3(a). Wet deposition loading of H+ (meq m−2 d−1) and gains or losses in the H+ loading of the snow cover at Lake Laflamme, spring 1983.

Fig.3.(b) Wet deposition loading of ![]() (meq m−2 d−1) and gains or losses in the
(meq m−2 d−1) and gains or losses in the ![]() loading of the snow cover at Lake Laflamme, spring 1983.
loading of the snow cover at Lake Laflamme, spring 1983.
Wet deposition, in the form of rain, was dominant during the 1983 melt season; from the onset of melt on 8 April to the disappearance of the pack on 15 May as much water fell from the atmosphere (rain: 19.2 cm, snowfall: water equivalent 2 cm) as was stocked originally (21 cm) in the pack on 8 April. These large amounts of wet deposition gave rise, according to the intensity and chemical content of the precipitation (eg: mean concentrations H+; 48.2 µeq 1−1, ![]() : 29.4 μeq 1−1,
: 29.4 μeq 1−1, ![]() : 18.26 µeq 1−1, K+ : 4 μeq 1−1,
: 18.26 µeq 1−1, K+ : 4 μeq 1−1, ![]() : BDL), to both losses and gains in the ionic loads of the snowpack. Although there was no consistent pattern of load losses and gains common to all the ionic species, gains were generally recorded during periods when snow fell or when rain filled up a drained pack close to the calculated saturation values for water retention (Reference AndersonAnderson 1976). Losses were noted during the pack by snow-melt or after rain events. In certain periods, losses were also recorded when continual rain and warm temperatures led to very high rates of meltwater discharge from the snow cover.
: BDL), to both losses and gains in the ionic loads of the snowpack. Although there was no consistent pattern of load losses and gains common to all the ionic species, gains were generally recorded during periods when snow fell or when rain filled up a drained pack close to the calculated saturation values for water retention (Reference AndersonAnderson 1976). Losses were noted during the pack by snow-melt or after rain events. In certain periods, losses were also recorded when continual rain and warm temperatures led to very high rates of meltwater discharge from the snow cover.
Mean values for the daily wet deposition loadings of ionic species associated with atmospheric aerosols (![]() : 0.32 ± 0.17,
: 0.32 ± 0.17, ![]() : 0.51 ± 0.34 meq m−2 d−1) were of approximately the same magnitude as the daily changes in gains (
: 0.51 ± 0.34 meq m−2 d−1) were of approximately the same magnitude as the daily changes in gains (![]() : 0.66 ± 0.47,
: 0.66 ± 0.47, ![]() : 0.62 ± 0.39 meq m−2 d−1) recorded in the snow cover during the melt period. In contrast, the mean value for the loading contribution by wet deposition to the total load of K+ (0.08 ± 0.04 meq m−2 d−1) (Fig.4(a)) in the snow cover was far outweighed by the gains (0.94 ± 0.82 meq m−2 d−1) which were recorded at this time. This observation was even more pronounced in the case of
: 0.62 ± 0.39 meq m−2 d−1) recorded in the snow cover during the melt period. In contrast, the mean value for the loading contribution by wet deposition to the total load of K+ (0.08 ± 0.04 meq m−2 d−1) (Fig.4(a)) in the snow cover was far outweighed by the gains (0.94 ± 0.82 meq m−2 d−1) which were recorded at this time. This observation was even more pronounced in the case of ![]() (Fig.4(b)). The very low concentrations of this ionic species in wet deposition were not reflected in the snow cover, where large gains in
(Fig.4(b)). The very low concentrations of this ionic species in wet deposition were not reflected in the snow cover, where large gains in ![]() loads (0.74 ± 1.05 meq m−2 d−1) comparable to those shown by
loads (0.74 ± 1.05 meq m−2 d−1) comparable to those shown by ![]() ,
, ![]() , K+ and H+ took place. K+ and
, K+ and H+ took place. K+ and ![]() production thus originated mainly from local phenomena within and/or on the snow cover while
production thus originated mainly from local phenomena within and/or on the snow cover while ![]() and
and ![]() gains were due mainly to atmospheric inputs.
gains were due mainly to atmospheric inputs.

Fig.4(a). Wet deposition loading of K+ (meq m−2 d−1) and gains or losses in the K+ loading of the snow cover at Lake Laflamme, spring 1983.

Fig.4(b). Wet deposition loading of ![]() (meq m−2 d−1) and gains or losses in the
(meq m−2 d−1) and gains or losses in the ![]() loading of the snow cover at Lake Laflamme, spring 1983.
loading of the snow cover at Lake Laflamme, spring 1983.
The evolution of H+ and ![]() loads in the pack showed similar behaviour during the end of the stable first phase (29 March–8 April) and during the second melt phase (8–16 April) (Figs. 3(a) and 3(b)). However, the subsequent overall relative increases and the concomitant patterns for losses and gains in the loads for the two ionic species in the pack differed considerably. As an example, a comparison of Figures 3(a) and 3(b) shows that on 17 April, when the
loads in the pack showed similar behaviour during the end of the stable first phase (29 March–8 April) and during the second melt phase (8–16 April) (Figs. 3(a) and 3(b)). However, the subsequent overall relative increases and the concomitant patterns for losses and gains in the loads for the two ionic species in the pack differed considerably. As an example, a comparison of Figures 3(a) and 3(b) shows that on 17 April, when the ![]() load in the pack increased in response to the wet deposition input for that day, the H+ load diminished in spite of a similar amount of wet deposition loading. An appreciable increase in K+ loading for this day largely compensated for the H+ loss in the ionic balance of the pack. Subsequently, the patterns for losses and gains in the snowpack for H+ and
load in the pack increased in response to the wet deposition input for that day, the H+ load diminished in spite of a similar amount of wet deposition loading. An appreciable increase in K+ loading for this day largely compensated for the H+ loss in the ionic balance of the pack. Subsequently, the patterns for losses and gains in the snowpack for H+ and ![]() were in opposition for periods in the fourth (25–30 April) and fifth (1–12 May) rain phases of the melt.
were in opposition for periods in the fourth (25–30 April) and fifth (1–12 May) rain phases of the melt.
In wet deposition, the concentrations and daily ionic loading of H+ was strongly correlated with that of ![]() (r2 = 0.85, n = 14, p < 0.001). No such correlation was found between H+ and
(r2 = 0.85, n = 14, p < 0.001). No such correlation was found between H+ and ![]() in the snow cores either before or during the snow melt period (Tables I and II). Although the sampling and analytical work-up of snow core samples (Reference JonesJones 1985) can contribute to the disappearance of the relationship found between these two ions in precipitation, the hydrogen ion also reacts to other autochtonous ionic transformations in the pack not common to precipitation chemistry. We suggest that the mechanisms responsible for losses and gains of other selected species such as the high K+ and
in the snow cores either before or during the snow melt period (Tables I and II). Although the sampling and analytical work-up of snow core samples (Reference JonesJones 1985) can contribute to the disappearance of the relationship found between these two ions in precipitation, the hydrogen ion also reacts to other autochtonous ionic transformations in the pack not common to precipitation chemistry. We suggest that the mechanisms responsible for losses and gains of other selected species such as the high K+ and ![]() loadings observed on 17 April can lead to the disappearance of atmospheric H+ and
loadings observed on 17 April can lead to the disappearance of atmospheric H+ and ![]() correlation coefficients in the pack. These ionic transformations within the pack are associated with organic matter and we further suggest that the main autochtonous mechanism for H+ suppression in boreal forest snow cover is that of ion exchange processes on organic material. This material may be either particulate or leached in soluble form from organic substrates during melt and rain periods. Organic matter may donate or accept protons depending depending on its structure, association with other ions (e.g. Ca2+, Mg2+, K+, Mn2+, Al3+) and biophysical context (Reference Lee and WeberLee and Weber 1982, Reference BacheBache 1983, Reference Krug and FrinkKrug and Frink 1983, Reference Driscoll and SchafranDriscoll and Schafran 1984). The presence of relatively large amounts of particulate organic matter in the pack is evident simply by direct observation. Although the presence of soluble organic matter in the snow cover was not measured directly during the snow melt of 1983, H G Jones and others (unpublished snow-melt data 1984) have noted net increases in the concentrations of soluble organic carbon in snowpacks and meltwaters over those recorded in the incident precipitation.
correlation coefficients in the pack. These ionic transformations within the pack are associated with organic matter and we further suggest that the main autochtonous mechanism for H+ suppression in boreal forest snow cover is that of ion exchange processes on organic material. This material may be either particulate or leached in soluble form from organic substrates during melt and rain periods. Organic matter may donate or accept protons depending depending on its structure, association with other ions (e.g. Ca2+, Mg2+, K+, Mn2+, Al3+) and biophysical context (Reference Lee and WeberLee and Weber 1982, Reference BacheBache 1983, Reference Krug and FrinkKrug and Frink 1983, Reference Driscoll and SchafranDriscoll and Schafran 1984). The presence of relatively large amounts of particulate organic matter in the pack is evident simply by direct observation. Although the presence of soluble organic matter in the snow cover was not measured directly during the snow melt of 1983, H G Jones and others (unpublished snow-melt data 1984) have noted net increases in the concentrations of soluble organic carbon in snowpacks and meltwaters over those recorded in the incident precipitation.
Table I Correlation Coefficients Between Data Relating the Concentrations of Chemical Species in Snow Cores Taken from the Snow Cover Within the Boreal Forest Site at Lake Laflamme in 1983 Between 15 March and 8 April (n = 59) and Between 15 March and 16 April ( n = 81) Inclusive

Table II Correlation Coefficients Between Data Relating the Concentrations of Chemical Species in Snow Cores Taken from the Snow Cover Within the Boreal Forest Site at Lake Laflamme in 1983 Between 17 April and 25 April (n = 35) and Between 26 April and 11 May (n = 44) Inclusive

Further indirect evidence for the presence or transformation of organic matter in the chemical evolution of snow cover is the correlation coefficients between data relating the concentrations of the ionic species K+, Ca2+, Mg2+ and Mn in the snow cores. Tables I and II show that, for all periods, K+, Ca2+, Mg2+ and Mn were strongly correlated (P ≤0.001). Factor analysis of the data also showed a regrouping that is indicative of a strong relationship between K+ Ca2+, Mg2+ and Mn (Fig.6). The factor relating the evolution of these chemical species together is their common source within the organic litter at the snow-cover level. These elements are strongly associated with vegetative tissue and are leached by precipitation in appreciable quantities from both detrital (e.g. Mn (Reference Tiffin, Mortvedt, Giordano and LindsayTiffin 1972)) and living matter such as forest canopies (K+, Mg2+ and Ca2+ (Reference Abrahamsen, Horntvedt and TveiteAbrahamsen and others 1977)).

Fig.6. Factor analysis of concentration data for ionic species in snow cores (n = 168) at Lake Laflamme during the spring season, 1983.
The expected losses for the snowpack, as calculated from the sum of the total loads stocked in the pack at the beginning of the melt periods and the total loadings in precipitation during the melt period, were lower than the sum of the actual losses observed for all ionic species except H+ (Table III). The increases (%) which occurred in the snowpack loads over and above that expected from the calculated losses during the melt may be regrouped into three groups (Table IV). The first grouping comprises the major anions Cl−, ![]() and
and ![]() , the increases of which were comparable (25 to 32%). These increases are net increases over the whole of the snowmelt period and, as such, cannot give any information as to the relative chemical or biochemical dynamics of the ionic species in the pack. As an example, we may point out that although Cl− and
, the increases of which were comparable (25 to 32%). These increases are net increases over the whole of the snowmelt period and, as such, cannot give any information as to the relative chemical or biochemical dynamics of the ionic species in the pack. As an example, we may point out that although Cl− and ![]() are often thought of as conservative ions (Reference Skartveit and GjessingSkartveit and Gjessing 1979), cyclic mechanisms of adsorption and desorption of these ions (particularly
are often thought of as conservative ions (Reference Skartveit and GjessingSkartveit and Gjessing 1979), cyclic mechanisms of adsorption and desorption of these ions (particularly ![]() ) in the pack could lead to the same net changes as those of a conservative ion. Organic matter in soils can retain
) in the pack could lead to the same net changes as those of a conservative ion. Organic matter in soils can retain ![]() by physical adsorption (Reference Johnson, Hendersen and ToddJohnson and others 1981) and the detrital organic matter in the pack may also have the capacity to adsorb this ion during certain periods. Although the net increases in loads do not give any information on chemical dynamics within the pack which could lead to a better understanding of ionic transformations and ton exchange processes in the snow cover, they do suggest, however, a common source for the increases in Cl−,
by physical adsorption (Reference Johnson, Hendersen and ToddJohnson and others 1981) and the detrital organic matter in the pack may also have the capacity to adsorb this ion during certain periods. Although the net increases in loads do not give any information on chemical dynamics within the pack which could lead to a better understanding of ionic transformations and ton exchange processes in the snow cover, they do suggest, however, a common source for the increases in Cl−, ![]() and
and ![]() . This would be dry deposition either directly by aerosol and gas interaction with the snow cover or indirectly by adsorption of the aerosols and gases on organic material followed by leaching during the melt period, or both. We believe that the latter phenomenon is probably the major source of these anions in the context of the boreal forest sites. The canopies of coniferous trees are scavengers of atmospheric aerosols (Reference Ibrahim, Barrie and FanakiIbrahim and others 1983) which are then removed by rain either from the canopy or from canopy material, which has been deposited on the snow.
. This would be dry deposition either directly by aerosol and gas interaction with the snow cover or indirectly by adsorption of the aerosols and gases on organic material followed by leaching during the melt period, or both. We believe that the latter phenomenon is probably the major source of these anions in the context of the boreal forest sites. The canopies of coniferous trees are scavengers of atmospheric aerosols (Reference Ibrahim, Barrie and FanakiIbrahim and others 1983) which are then removed by rain either from the canopy or from canopy material, which has been deposited on the snow.
Table III Chemical Budgets for Snow-Cover Total Loading Losses (meq m−2) During the Melt Season at Lake Laflamme, 1983

Table IV Increases (%) or Decreases (−) of Measured Ionic Loading Losses from the Snow Cover Over those Calculated from Precipitation Loadings and Original Snow Cover Loading Before the Spring Melt


Fig.5. Relationship between changes in anionic and cationic loadings (meq m−2 d−1) (losses or gains) in the snow cover at Lake Laflamme, 1983.
The second group of ionic species is composed of Ca2+ , ![]() , Mg2+ and Na+; the values for the increases in loads for these ionic species (50 to 287%) may represent, in addition to leaching of local debris, the exudates of cellular material from the cell plasmolysis of detrital organic debris (particularly Ca2+, Mg2+ and Na+), In the case of
, Mg2+ and Na+; the values for the increases in loads for these ionic species (50 to 287%) may represent, in addition to leaching of local debris, the exudates of cellular material from the cell plasmolysis of detrital organic debris (particularly Ca2+, Mg2+ and Na+), In the case of ![]() , microbiological activity of a heterotrophic nature within the pack (i.e. snow fungi, bacteria) is also a factor to be considered (Reference Stein and AmundsenStein and Amundsen 1967, Reference VisserVisser 1973). High rates of production within the pack, however, are characteristic of the third group of ionic species (Al3+, Mn2+, K+ and
, microbiological activity of a heterotrophic nature within the pack (i.e. snow fungi, bacteria) is also a factor to be considered (Reference Stein and AmundsenStein and Amundsen 1967, Reference VisserVisser 1973). High rates of production within the pack, however, are characteristic of the third group of ionic species (Al3+, Mn2+, K+ and ![]() ) which show substantial increases in pack loads (480 to 750%). These increases cannot be accounted for by any other local phenomena other than the dissolution and/or microbiological degradation of organic debris.
) which show substantial increases in pack loads (480 to 750%). These increases cannot be accounted for by any other local phenomena other than the dissolution and/or microbiological degradation of organic debris.
The presence of ![]() is particularly indicative of heterotrophic microbiological activity or secondary productivity of organic matter at higher levels. K+ and
is particularly indicative of heterotrophic microbiological activity or secondary productivity of organic matter at higher levels. K+ and ![]() may be released into the pack by dissolution subsequent to microbiological degradation of organic material, An alternate source may be macrobiological excretion of
may be released into the pack by dissolution subsequent to microbiological degradation of organic material, An alternate source may be macrobiological excretion of ![]() by the high populations of Collembola (Reference HumbertHumbert 1980) present in the pack in spring time at Lake Laflamme.
by the high populations of Collembola (Reference HumbertHumbert 1980) present in the pack in spring time at Lake Laflamme. ![]() is a strong acid anion and could, at the ionic loadings recorded, have compensated in part for the proposed mechanism of H+ removal in the pack.
is a strong acid anion and could, at the ionic loadings recorded, have compensated in part for the proposed mechanism of H+ removal in the pack.
One must, however, be cautious in arriving at any conclusions that may be drawn from this type of dat analysis in snow-cover studies. Certain paradoxes exist, which may or may not be related to the difficulties inherent in the sampling of such a heterogeneous milieu, in the treatment of the samples during analytical techniques (Reference JonesJones 1985) and even in the expressions used to calculate mean values for concentrations or loads of ionic species in the pack. For example, the water-equivalent budget (38.29 cm) for the snow cover calculated from the sum of periodic losses of water equivalent in the declining pack is very similar to the water equivalent budget (38.54 cm) calculated from the initial mean water equivalent of the pack before the spring melt and the water equivalent of precipitation inputs. As outlined above, the method used for the calculation of ionic loadings from periodic losses leads to a total loss in the load of ![]() which is 25% higher than that expected from the calculated loadings from precipitation. As the load in the pack has increased but the water equivalent has not, one would expect to find higher mean concentrations of
which is 25% higher than that expected from the calculated loadings from precipitation. As the load in the pack has increased but the water equivalent has not, one would expect to find higher mean concentrations of ![]() in the snow cover than in the precipitation. This was, however, not observed. The lower mean (arithmetic) values of
in the snow cover than in the precipitation. This was, however, not observed. The lower mean (arithmetic) values of ![]() in the snow cores compared to mean
in the snow cores compared to mean ![]() concentrations in the precipitation for the snowmelt period may be an artifact due to the large variability of
concentrations in the precipitation for the snowmelt period may be an artifact due to the large variability of ![]() concentrations observed in the snow cover and the method used for the calculation of
concentrations observed in the snow cover and the method used for the calculation of ![]() load.
load.
In conclusion, the results suggest that organic matter plays a major role in the evolution of the chemical load of many ionic species in the snow cover of boreal forest sites. The ion exchange capacity of both particulate and soluble organic material leads to a decrease in pack acidity and large increases in the loadings of essential nutrients and micronutrients such as K2+, ![]() and Mn. These in-pack transformations then destroy the relationship between concentrations of (H+) and the concentrations of strong acid anions (
and Mn. These in-pack transformations then destroy the relationship between concentrations of (H+) and the concentrations of strong acid anions (![]() ,
, ![]() ) that are found in precipitation as it is deposited from the atmosphere. The influence of organic matter on snowpack chemistry should thus be taken into consideration in any attempt to model acid rain fluxes through boreal forest systems.
) that are found in precipitation as it is deposited from the atmosphere. The influence of organic matter on snowpack chemistry should thus be taken into consideration in any attempt to model acid rain fluxes through boreal forest systems.












