


Exclusive breast-feeding has long been associated with lower morbidity and mortality during the first year of life, as well as with reduced incidence and severity of infections and immune-related diseases, compared with formula feeding( 1 ). Breast milk not only provides essential nutrients and contains a broad range of bioactive, immunological and protective compounds unequalled by current formulae( 1 ), but has moreover been recognised as a continuous source of commensal and potential probiotic bacteria, able to influence initial neonatal gut colonisation and maturation of the immune system( Reference Fernandez, Langa and Martin 2 ). Thus, understanding the impact of breast milk on early host–microbe interactions may provide future opportunities for modulating a dysbiotic microbiota and reduce the risk of disease( Reference Rautava, Luoto and Salminen 3 ).
Predominant culturable bacterial populations in breast milk have been identified as facultative anaerobic members of the Bacilli (Firmicutes phylum), i.e. Staphylococcus and Streptococcus spp.( Reference Heikkila and Saris 4 , Reference Solis, de Los Reyes-Gavilan and Fernandez 5 ). At the subdominant level, a range of facultative anaerobic strains have been isolated, including members of the Actinobacteria phylum (e.g. Propionibacterium and Rothia spp.)( Reference Heikkila and Saris 4 , Reference Jimenez, Delgado and Maldonado 6 ), members of the Bacilli, comprising lactic acid bacteria (e.g. Enterococcus and Lactobacillus spp.)( Reference Solis, de Los Reyes-Gavilan and Fernandez 5 , Reference Albesharat, Ehrmann and Korakli 7 ), and occasionally members of the Enterobacteriaceae (Proteobacteria phylum)( Reference Jimenez, Delgado and Maldonado 6 , Reference Perez, Dore and Leclerc 8 ). Because many culture-dependent studies were conducted in view of isolating potential probiotics, breast milk was furthermore shown to harbour obligate anaerobic gut-associated Bifidobacterium spp.( Reference Solis, de Los Reyes-Gavilan and Fernandez 5 , Reference Jimenez, Delgado and Maldonado 6 , Reference Arboleya, Ruas-Madiedo and Margolles 9 – Reference Makino, Kushiro and Ishikawa 12 ), which besides Bacteroides spp. represent the major anaerobic population of early gut microbiota in breast-fed neonates( Reference Jost, Lacroix and Braegger 13 ). However, until now, little work has been performed on the detection of other viable major gut-associated anaerobes in breast milk, i.e. members of the Bacteroidetes phylum and the Clostridia class (Firmicutes phylum).
Culture-independent methods have moreover begun to reveal the presence of DNA belonging to gut-associated anaerobic bacterial taxa not detected previously in breast milk in culture-dependent studies( Reference Perez, Dore and Leclerc 8 , Reference Collado, Delgado and Maldonado 14 – Reference Hunt, Foster and Forney 17 ). PCR–denaturing gradient gel electrophoresis, cloning and 16S ribosomal RNA (rRNA) sequencing have led to the detection of the genera Bacteroides, Clostridium, Eubacterium and Veillonella in breast milk( Reference Perez, Dore and Leclerc 8 , Reference Martin, Heilig and Zoetendal 15 ). In addition, quantitative PCR analyses allowed for the quantification of the Bacteroides group and different groups of the Clostridia, including the Clostridium clusters IV and XIV that comprise the major butyrate producers important for colonic health (i.e. Faecalibacterium and Roseburia, respectively)( Reference Collado, Delgado and Maldonado 14 , Reference Cabrera-Rubio, Collado and Laitinen 16 ). Recently, high-throughput sequencing (i.e. 454-pyrosequencing) has been carried out on breast milk samples collected from 1 to 10 months postpartum, which allowed gaining the broadest insight into bacterial diversity in breast milk( Reference Cabrera-Rubio, Collado and Laitinen 16 , Reference Hunt, Foster and Forney 17 ). In both studies, facultative anaerobic members of the Bacilli accounted for the majority of DNA pyrosequences, but at a lower taxonomic level, large differences in relative abundance were apparent. While Staphylococcus and Streptococcus were predominant in the study by Hunt et al. ( Reference Hunt, Foster and Forney 17 ), which is supported by culture-dependent research( Reference Heikkila and Saris 4 , Reference Solis, de Los Reyes-Gavilan and Fernandez 5 ), in the study by Cabrera-Rubio et al. ( Reference Cabrera-Rubio, Collado and Laitinen 16 ), Weissella and Leuconostoc predominated over these genera. Despite these differences, which may be due to geographical and/or methodological factors, in both studies, sequences belonging to the obligate anaerobic genera Prevotella (Bacteroidetes phylum) and Veillonella (Clostridia class) were detected. However, although some obligate gut-associated anaerobes have been detected in breast milk using culture-independent methods, no information about the viability of these populations could be gained. Thus, without confirming the presence of such members of the Bacteroidetes and Clostridia by culture and isolation of corresponding strains, it remains unclear whether breast milk is a source of such gut-associated anaerobes influencing initial gut colonisation, or whether dead cells or parts thereof are transferred to the breast-fed neonate. Therefore, the purpose of the present study was to investigate bacterial diversity in breast milk, emphasising on the identification of gut-associated anaerobes by combining aerobic and anaerobic culture methods with state-of-the-art, culture-independent methods, including Sanger sequencing and high-throughput sequencing (i.e. 454-pyrosequencing).
Experimental methods
Subjects, study design and sample collection
Healthy mothers-to-be, carrying a healthy baby and planning to deliver vaginally at term and to exclusively breast-feed during the postpartum period, were recruited for the present observational clinical study at the University Children's Hospital Zurich and the Hospital Zollikerberg (Zurich, Switzerland). Exclusion criteria were preterm and/or caesarean delivery, any formula feeding in addition to breast-feeding, as well any variables known to affect the balance of maternal (gut) microbiota, such as gastrointestinal and immunological disorders, and drug administration (e.g. antibiotics, laxatives) during the neonatal period and at least 4 months before delivery. The present study was conducted according to the guidelines laid down in the Declaration of Helsinki, and all procedures involving human subjects were approved by the Ethics Committee of the Canton of Zurich (Zurich, Switzerland; project identification code: StV 47/08; date of approval: 12 October 2009). Written informed consent was obtained from all subjects.
Breast milk samples were collected from seven mothers at three sampling points, i.e. at days 3–6, 9–14 and 25–30 postpartum. Due to the stringent inclusion criteria, two mothers were excluded from the study after the first sampling point and another twelve prepartum, mainly after caesarean delivery and/or antibiotic treatment. After rejection of foremilk and cleaning of the breast with aseptic soap, milk was collected using a hospital-grade electrical breast pump sterilised by autoclaving (Symphony). To minimise the exposure to O2, 5–15 ml of breast milk were injected with a sterile syringe into a rubber-stoppered 100 ml vessel devoid of air by flushing with CO2 before sterilisation. Samples were transported at 4°C and processed within 4 h in an anaerobic chamber (Coy Laboratory Products, Inc.) with an atmosphere of 85 % N2, 10 % CO2 and 5 % H2 (PanGas AG). Aliquots of 2 ml were immediately subjected to culture, while further aliquots were stored at − 80°C before DNA extraction and 454-pyrosequencing analysis.
Culture and isolation
For the quantification of viable bacterial populations in breast milk and the subsequent isolation of strains, aliquots of 100 μl were plated in duplicate on two non-selective and seven selective agar media. The following media targeting anaerobic, major gut-associated bacterial populations were incubated in an anaerobic chamber: Bacteroides mineral salts agar for Bacteroides spp. (using 5 g/l of d-glucose as the carbon source; VWR International)( Reference Macfarlane, Hay and Macfarlane 18 ), Beerens agar for Bifidobacterium spp.( Reference Beerens 19 ), reinforced clostridial agar for members of the Clostridia( Reference Steer, Gee and Johnson 20 ) and Wilkins–Chalgren anaerobe agar (WC) for total anaerobes (Oxoid AG; supplemented with 0·5 g/l of l-cysteine–HCl; Sigma-Aldrich). On the other hand, media targeting facultative anaerobic populations, i.e. MacConkey agar no. 2 (MC) for Enterobacteriaceae/Enterococcus spp. (Oxoid), mannitol salt agar for Staphylococcus spp. (Oxoid) and nutrient agar for total facultative anaerobes (Oxoid), were incubated aerobically. The Lactobacillus anaerobic de Man, Rogosa and Sharpe agar with vancomycin and bromocresol green (LAMVAB) targeting Lactobacillus spp.( Reference Hartemink, Domenech and Rombouts 21 ) and azide blood agar for gram-positive cocci/Streptococcus spp. (Oxoid) were incubated in anaerobic jars. All plates were incubated at 37°C for 2–14 d and population levels are reported as log colony-forming units (cfu)/ml breast milk.
Based on different morphologies, a set of colonies was isolated per sample and agar medium, streaked for purity and cultured in liquid media, i.e. Wilkins–Chalgren anaerobe broth for presumptive anaerobes (Oxoid; supplemented with 0·5 g/l of l-cysteine–HCl; Sigma-Aldrich), tryptone soya broth for facultative anaerobes (Oxoid) and de Man, Rogosa and Sharpe broth for presumptive Lactobacillus spp. (Labo-Life Sàrl; supplemented with 0·5 g/l of l-cysteine–HCl; Sigma-Aldrich). Purity was verified microscopically and viable isolates were maintained at − 80°C in 20 % (v/v) glycerol, while in addition, cell pellets obtained by centrifugation (8000 g , 10 min) were stored at − 20°C before DNA extraction and Sanger sequencing.
DNA extraction
DNA was extracted from pure culture cell pellets for subsequent Sanger sequencing using a Wizard Genomic DNA purification kit (Promega AG) according to the manufacturer's instructions. For pyrosequencing analysis, total DNA was extracted from pellets obtained by centrifugation (8000 g , 20 min) of 1 ml aliquots of breast milk, which were resuspended in sodium phosphate buffer (MP Biomedicals), using a FastDNA SPIN Kit for Soil and the FastPrep-24 instrument for mechanical lysis (MP Biomedicals) according to the manufacturer's instructions. DNA concentration and quality were assessed spectrophotometrically (NanoDrop 1000; Witec AG) and stored at − 20°C before molecular analyses.
Sanger sequencing
PCR amplification of near full-length 16S rRNA genes was performed using a 4:1 mixture of forward primers 8f (5′-AGAGTTTGATCMTGGCTCAG-3′, universal) and 8f-bif (5′-AGGGTTCGATTCTGGCTCAG-3′, Bifidobacterium-specific) and a universal bacterial reverse primer 1391R (5′-GACGGGCGGTGTGTRCA-3′) (Microsynth AG) as described previously( Reference Dethlefsen, Huse and Sogin 22 ). Reactions of 50 μl contained 25 μl of 2 × MasterMix (Fermentas GmbH), 0·2 μm of each primer (-mixture) and 1 μl of template DNA diluted to 1 ng/μl. Thermocycling (Biometra TProfessional Thermocycler; Biolabo Scientific Instruments SA) was performed with an initial denaturation step at 94°C for 300 s, followed by thirty cycles of denaturation at 94°C for 30 s, annealing at 57°C for 60 s and extension at 72°C for 30 s and a final extension at 72°C for 420 s. Specificity and amplicon size were verified by electrophoresis in 1·5 % (w/v) agarose gels, and reactions were purified using an illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare Europe GmbH) according to the manufacturer's instructions.
Cycle sequencing PCR was carried out in 20 μl reaction volumes with 5 % (v/v) BigDye v3·1 (Applied Biosystems Europe BV), 4 μl of 5 × sequencing buffer (Applied Biosystems), 1 μm of reverse primer 1391R and 1 μl of purified PCR template. Thermocycling (labcycler; SensoQuest GmbH) was performed with an initial denaturation step at 96°C for 300 s, followed by thirty-five cycles of denaturation at 96°C for 10 s, annealing at 55°C for 20 s and extension at 60°C for 240 s. Reactions were purified by dextran gel bead filtration (Sephadex; GE Healthcare) before loading 10 μl for capillary electrophoresis (ABI 3130xl DNA Analyzer; Applied Biosystems). Sequencing trace chromatograms were quality-trimmed and checked for miscalled bases using a chromatogram viewer (FinchTV v1.4.0; Geospizia, Inc.). The Basic Local Alignment Search Tool algorithm( Reference Altschul, Gish and Miller 23 ) was used to align sequences with the GenBank database( Reference Benson, Karsch-Mizrachi and Lipman 24 ), and phylogenetic assignments were based on the nearest neighbour ( ≥ 97 % sequence similarity), excluding sequences deposited from uncultured samples.
Pyrosequencing
High-throughput sequencing of total DNA extracted from breast milk was carried out at DNAVision SA (Charleroi, Belgium) using a 454 Life Sciences system in combination with Titanium chemistry (Roche AG).
Partial 16S rRNA genes were amplified by PCR using a forward primer containing the Titanium A adaptor sequence (5′-CCATCTCATCCCTGCGTGTCTCCGACTCAG-3′), a 5–10 nt multiplex identifier sequence and a template-specific primer sequence. The reverse primer contained the Titanium B adaptor sequence (5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAG-3′) and a template-specific primer sequence. Template-specific primer sequences (5′-AGGATTAGATACCCTGGTA-3′ and 5′-CRRCACGAGCTGACGAC-3′) allowed targeting the V5–V6 hypervariable 16S rRNA region( Reference Andersson, Lindberg and Jakobsson 25 ). Each reaction mixture of 100 μl contained 20 μl of 5 × KAPA HiFi Fidelity buffer, 2U of KAPA HiFi Hotstart DNA polymerase, 0·3 mm of each deoxyribonucleotide triphosphate (Kapa Biosystems), 0·3 μm of each primer (Eurogentec) and 60 ng of template DNA. Thermocycling was performed with an initial denaturation step at 95°C for 5 min, followed by twenty-five cycles of denaturation at 98°C for 20 s, annealing at 56°C for 40 s and extension at 72°C for 20 s, with a final extension of 5 min at 72°C. Specificity and amplicon size were verified by electrophoresis in 1 % (w/v) agarose gels, and amplicons were purified using a Wizard SV Gel and PCR Clean-up System (Promega) according to the manufacturer's instructions.
Amplicons were quantified using a Quant-iT PicoGreen dsDNA assay kit (Life Technologies Corporation) according to the manufacturer's instructions and combined in equimolar concentrations for multiplexing. The final pool of DNA was purified using an Agencourt AMPure XP system (Agencourt Bioscience Corporation), according to the manufacturer's instructions, and resuspended in 100 μl of Tris–EDTA buffer. Unidirectional pyrosequencing was then carried out using Primer A on a 454 Life Sciences Genome Sequencer GS FLX instrument (Roche) following Titanium chemistry.
Sequence quality was then verified according to the criteria: a maximum of one mismatch in the barcode and primer, a length of at least 240 nt and a maximum of two undetermined bases per sequence (excluding barcode and primers). Phylum-, family- and genus-level taxonomic assignments of sequences that passed quality control were made using the Ribosomal Database Project Classifier (version 2.1; Center for Microbial Ecology, Michigan State University; http://rdp.cme.msu.edu/)( Reference Wang, Garrity and Tiedje 26 ) with a confidence threshold of 80 %. The Mothur software package (Department of Microbiology and Immunology, University of Michigan; http://www.mothur.org/) was used for nearest-neighbour clustering of the sequences into operational taxonomic units, based on which Chao1 richness and Shannon diversity estimations were calculated( Reference Schloss, Westcott and Ryabin 27 , Reference Chao and Shen 28 ).
Statistical analysis
Quantitative data were obtained from culture (duplicates), averaged and log10-transformed. Mean log-transformed values and relative abundance data from pyrosequencing were calculated at each time point (means and standard deviations). Means were compared using Student's t test and non-parametric Wilcoxon–Kruskal–Wallis tests with significance levels of P< 0·05 using JMP statistical software (version 9; SAS Institute, Inc.).
Results
Culture, isolation and strain typing
Breast milk harboured low mean viable bacterial counts < log 3 cfu/ml, as detected on the non-selective agar media, WC and nutrient agar, targeting total anaerobes (log 2·5 (sd 0·9) cfu/ml; range log 1·1–4·4 cfu/ml) and total facultative anaerobes (log 2·1 (sd 1·1) cfu/ml; range log 0–4·2 cfu/ml), respectively. Culture on seven selective media, however, revealed the predominance of presumptive Staphylococcus and Streptococcus populations, and low levels of presumptive Bifidobacterium, Enterobacteriaceae/Enterococcus and Clostridia populations, whereas no presumptive Bacteroides or Lactobacillus populations were detected on the respective selective agar media (Fig. 1). No significant trends of changes of bacterial population densities were detected within the first month postpartum (results not shown).
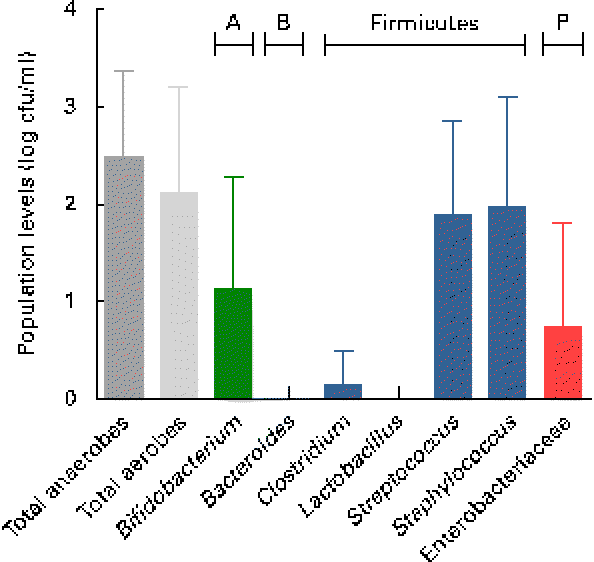
Fig. 1 Presumptive bacterial populations detected in breast milk collected from seven mothers at three sampling points, using culture on non-selective and selective agar media. Values are means, with standard deviations represented by vertical bars (n 21). A, Actinobacteria; B, Bacteroidetes; P, Proteobacteria; cfu, colony-forming units.
Following quantification of viable populations in each of the three breast milk samples obtained from the seven mothers, a total of 233 strains were isolated from the different agar media (sixty-two to eighty-six isolates per time point) based on different morphologies. Bacterial isolates were subjected to 16S rRNA gene sequencing, which after quality trimming resulted in partial sequences of 712 (sd 212) bp. Alignment of the sequences with those deposited in the GenBank database revealed that the majority of strains isolated from breast milk were members of the Firmicutes and Actinobacteria phyla, while members of Proteobacteria or Bacteroidetes were few or not detected, respectively. Over 90 % of the strains isolated from breast milk belonged to three facultative anaerobic genera, Staphylococcus (60·1 %), Streptococcus (17·6 %) and Propionibacterium (13·7 %). More specifically, at the species level, Staphylococcus epidermidis, Streptococcus salivarius/thermophilus/vestibularis and Propionibacterium acnes strains were isolated from the breast milk samples of all seven mothers. An overview of the taxonomic assignment of the isolated strains, their isolation frequency in relation to the total amount of isolates, as well as subject-specific prevalence are given in Table 1.
Table 1 Taxonomy of strains (n 233) isolated from breast milk of seven mothers, based on closest relatives (≥97 % sequence similarity) using 16S ribosomal RNA gene sequence alignments with GenBank
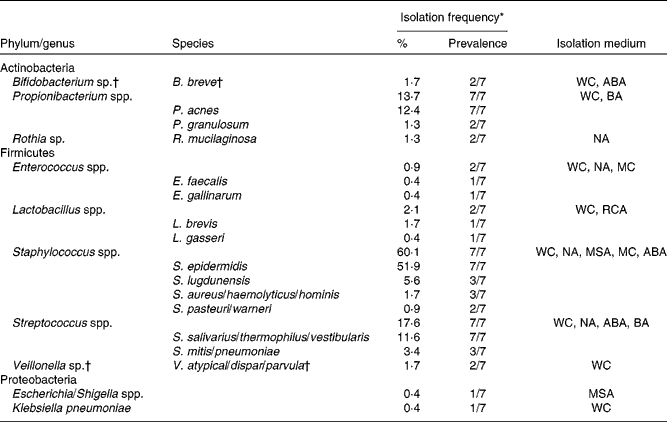
WC, Wilkins–Chalgren anaerobe agar; ABA, azide blood agar; BA, Beerens agar; NA, nutrient agar; MC, MacConkey agar no. 2; RCA, reinforced clostridial agar; MSA, mannitol salt agar.
* Percentage of the total number of isolates (n 233); prevalence refers to the number of subjects from which the corresponding taxa were isolated on at least one occasion.
† Obligate anaerobic taxa.
As revealed by the partial 16S rRNA sequencing of isolates, the selective culture media did not always support the growth of the respective target populations. For instance, although presumptive Staphylococcus and Streptococcus populations were quantified at equally high levels (Fig. 1), isolates from azide blood agar comprised not only the strains of Streptococcus, but also the strains of Staphylococcus and Bifidobacterium. Likewise, the presumptive Enterobacteriaceae/Enterococcus population detected with MC agar was overestimated (Fig. 1), as beyond expectations, the growth of Staphylococcus spp. was not inhibited on this medium. No growth was detected on the highly selective LAMVAB agar targeting Lactobacillus spp.; however, few strains of Lactobacillus were isolated from both WC and reinforced clostridial agar agars. Similarly, strains isolated from Beerens agar targeting Bifidobacterium spp. were identified as Propionibacterium and Streptococcus spp., but strains of Bifidobacterium were isolated from the non-selective WC agar. Furthermore, few strains belonging to the genus Rothia, the opportunistic pathogen Klebsiella, as well as of the obligate anaerobic, lactate utiliser Veillonella, were isolated from the non-selective WC agar.
Pyrosequencing
High-throughput sequencing performed on breast milk DNA generated a total of 106 075 quality-filtered, taxonomically classifiable 16S rRNA gene sequence reads with a mean of 5051 (sd 1523) reads per sample and a mean read length of 258 (sd 1) bp. The mean number of observed operational taxonomic units was 512 (sd 237) at 3 % similarity cut-off, based on which richness (Chao1 index: 1257 (sd 613)) and diversity (non-parametric Shannon index: 3·73 (sd 1·05)) were estimated.
Using the Ribosomal Database Project Classifier, sequences were assigned to the closest related taxa with a similarity threshold of 80 %. Mean relative abundances at the phylum level for each of the three breast milk sampling points revealed the predominance of four major phyla (Fig. 2). The Firmicutes and Proteobacteria phyla were the most abundant (approximately 42 and 35 %, respectively), followed by the Actinobacteria and Bacteroidetes phyla (approximately 9 and 11 %, respectively), while, furthermore, the Acidobacteria phylum was detected at subdominant levels ( < 1 %) (Fig. 2(a)).
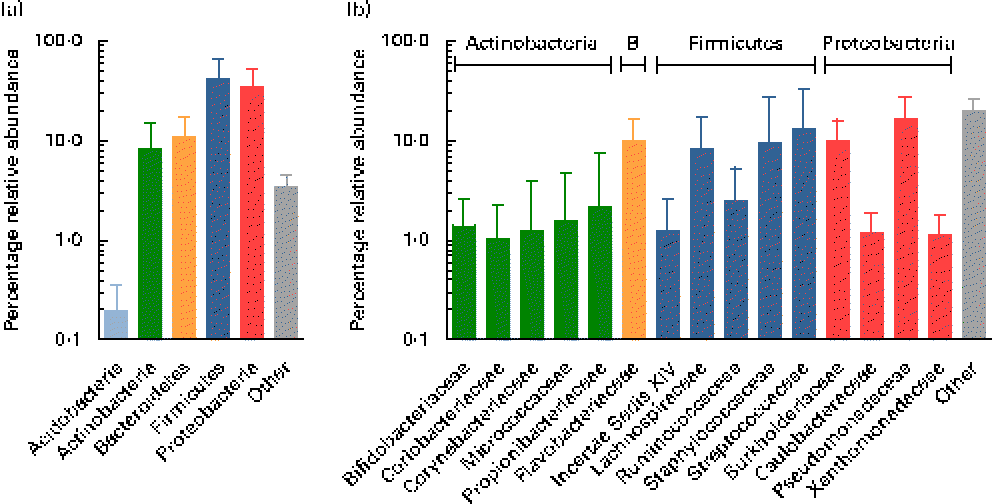
Fig. 2 Mean relative 16S ribosomal RNA gene abundances detected in breast milk at (a) the phylum level and (b) the family level (>1 % mean relative abundance), using pyrosequencing. Values are means for all seven mothers at all three sampling points, with standard deviations represented by vertical bars (n 21). B, Bacteroidetes.
At the family level, the most abundant phylum, Firmicutes, was composed of Streptococcaceae and Staphylococcaceae (approximately 13 and 10 %, respectively), i.e. families whose members are facultative anaerobes. However, surprisingly, families comprising obligate anaerobic, gut-associated members were represented at relatively high relative abundances, i.e. Lachnospiraceae, Ruminococcaceae, as well as members of the Clostridiales Incertae Sedis XIV (Fig. 2(b)). The second most abundant phylum, Proteobacteria, comprised members of the four families, mainly Pseudomonadaceae and Burkholderiaceae (approximately 17 and 10 %, respectively), while Caulobacteraceae and Xanthomonadaceae were detected at lower relative abundances ( < 2 %). Flavobacteriaceae was the only family detected at high relative abundance within the phylum Bacteroidetes (approximately 10 %). The phylum Actinobacteria was largely composed of Propionibacteriaceae, Bifidobacteriaceae, Coriobacteriaceae, Corynebacteriaceae and Micrococcaceae, each at relative abundances ≤ 2 %.
At the genus level, at total of 193 genera were detectable (i.e. >0·01 % relative abundance) in at least one sample. However, twelve genera appeared to be predominant in the breast milk ecosystem, as they were detected in ≥ 90 % of individual samples with a mean relative abundance >1 %, and observed in the samples of all seven mothers (except for Rothia, which was detected in six of the seven mothers and fifteen of the twenty-one samples). The relative abundances of these predominant genera were individual-specific and subject to intra-individual variations over time, such as the large fluctuations of Streptococcus and Staphylococcus over time observed in the profiles of subjects n8 and n12, respectively (Fig. 3).
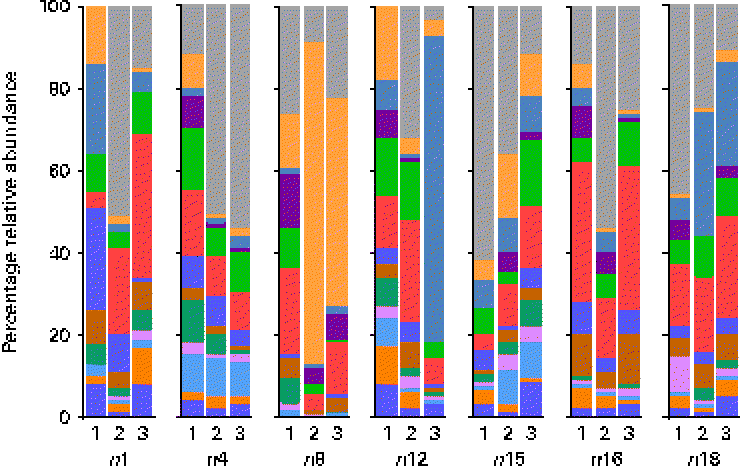
Fig. 3 Relative 16S ribosomal RNA gene abundances of the twelve predominant genera detected in ≥ 90 % of individual samples (n 21) at a mean relative abundance >1 %, and observed in at least one of the three breast milk samples collected from each of the seven mothers (except six of the seven mothers for Rothia, fifteen of the twenty-one samples), using pyrosequencing. ![]() , Bifidobacterium;
, Bifidobacterium; ![]() , Blautia;
, Blautia; ![]() , Brevundimonas;
, Brevundimonas; ![]() , Burkholderia;
, Burkholderia; ![]() , Corynebacterium;
, Corynebacterium; ![]() , Flavobacterium;
, Flavobacterium; ![]() , Propionibacterium;
, Propionibacterium; ![]() , Pseudomonas;
, Pseudomonas; ![]() , Ralstonia;
, Ralstonia; ![]() , Rothia;
, Rothia; ![]() , Staphylococcus;
, Staphylococcus; ![]() , Streptococcous;
, Streptococcous; ![]() , other. Indexes 1, 2 and 3, sampling points at days 3–6, 9–14 and 25–30 postpartum, respectively.
, other. Indexes 1, 2 and 3, sampling points at days 3–6, 9–14 and 25–30 postpartum, respectively.
Overall, the highest mean relative abundance was observed for the genus Pseudomonas (approximately 17 %), followed by the genera Streptococcus, Staphylococcus and Ralstonia, each at a mean relative abundance between 8 and 13 % (Fig. 4(a)). While the high relative abundances of Streptococcus and Staphylococcus confirm the results of the culture-dependent approach, no strains of Pseudomonas or Ralstonia (previously included in the genus Pseudomonas) were isolated by culture, although MC agar medium is expected to support their growth. Furthermore, the presence of Bifidobacterium, Propionibacterium as well as Rothia at lower relative abundances is well supported by culture and isolation of the respective genera (Table 1). In contrast, although few Lactobacillus strains were isolated by culture, this genus was detected at a mean relative abundance below 0·1 % only.
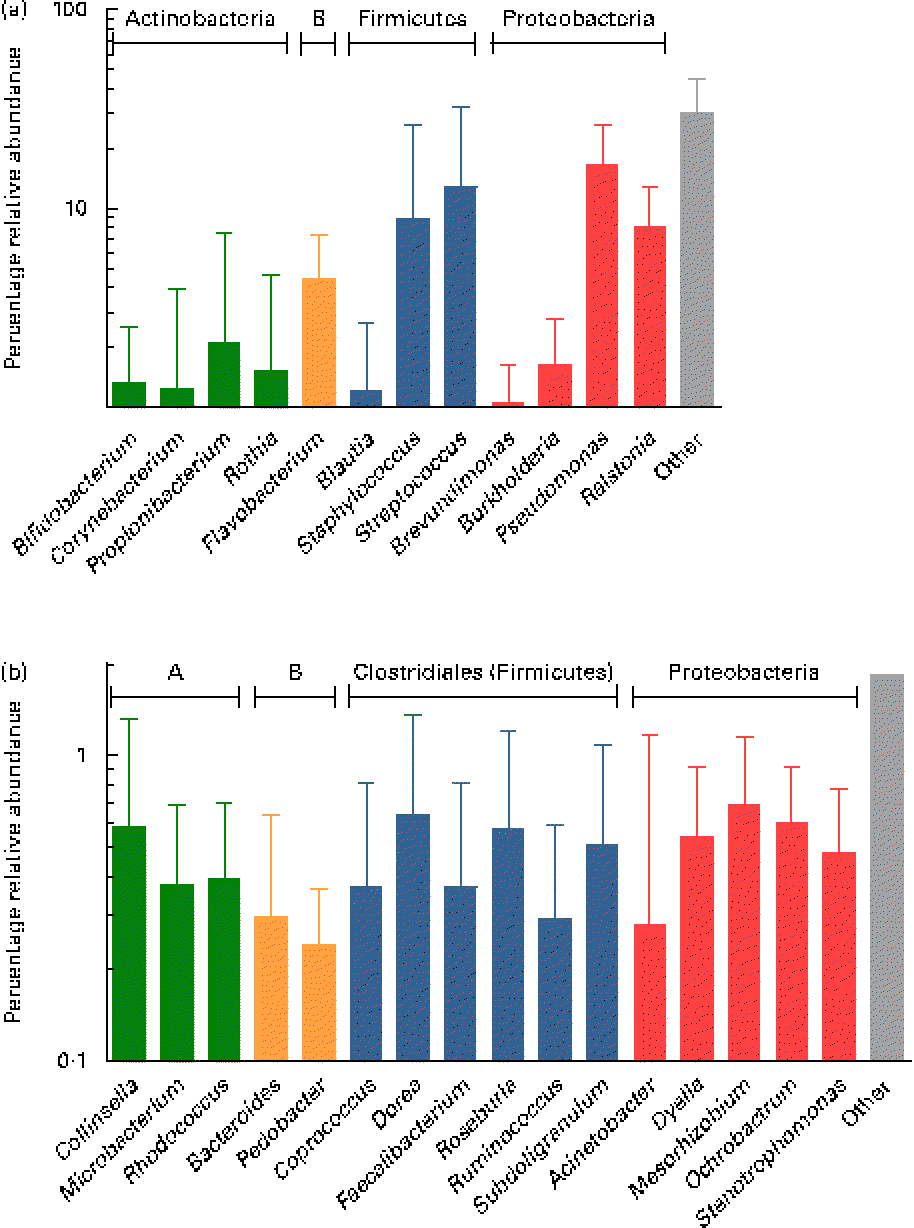
Fig. 4 (a) Mean relative 16S ribosomal RNA gene abundances of the twelve predominant genera detected in ≥ 90 % of individual samples at a mean relative abundance >1 %, and observed in at least two of the three breast milk samples from each of the seven mothers (except six of the seven mothers for Rothia, fifteen of the twenty-one samples), using pyrosequencing. (b) Mean relative 16S ribosomal RNA gene abundances of the sixteen subdominant genera detected at a mean relative abundance between 0·1 and 1 % and present in at least one breast milk sample from each mother, and in more than two of the three individual samples. Anaerobes: (a) Bifidobacterium and Blautia; (b) Collinsella, Bacteroides, Coprococcus, Dorea, Faecalibacterium, Roseburia, Ruminococcus and Subdoligranulum. Butyrate producers: Coprococcus, Faecalibacterium, Roseburia and Subdoligranulum. Values are means for all seven mothers at all three sampling points, with standard deviations represented by vertical bars (n 21). A, Actinobacteria; B, Bacteroidetes.
While, using culture, Veillonella spp. were identified as gut-associated obligate anaerobes belonging to the Clostridia class in the samples of two mothers, this genus was also detected at low abundance in four of the twenty-one samples from three mothers using pyrosequencing. Furthermore, the anaerobic genus Blautia (Clostridia class) was detected in nineteen of the twenty-one samples at a mean relative abundance of 1·2 % (Fig. 4(a)). At the subdominant level (0·11 % mean relative abundance), sixteen genera were observed in at least one breast milk sample of each mother, and in sixteen or more of the twenty-one individual samples. The identified genera comprised typical gut-associated obligate anaerobes (6·4 % relative abundance), such as Bacteroides and multiple members of the Clostridia, including besides the genera Dorea and Ruminococcus, major butyrate producers, such as Faecalibacterium and Roseburia, as well as Coprococcus and Subdoligranulum (Fig. 4(b)).
Discussion
The aim of the present study was to investigate whether breast milk represents a source of bacterial diversity, including major gut-associated anaerobes such as members of Bacteroidetes and Clostridia, influencing the initial gut colonisation of the breast-fed neonate. Therefore, aerobic and anaerobic culture methods were successfully combined with state-of-the-art, culture-independent methods, including Sanger sequencing and high-throughput sequencing (i.e. 454-pyrosequencing).
Culture on non-selective agar media revealed that breast milk harboured low mean viable bacterial counts < log 3 cfu/ml, which is in agreement with previous reports using culture-dependent methods( Reference Heikkila and Saris 4 , Reference Perez, Dore and Leclerc 8 ). However, Solis et al. ( Reference Solis, de Los Reyes-Gavilan and Fernandez 5 ) reported mean values of log 3–5 cfu/ml for samples from healthy mothers, which may be attributed to differences in culture conditions and/or to geographical factors. Furthermore, the authors have observed decreasing bacterial densities in breast milk over time, while this was not confirmed in the present study, where no decreasing or increasing trend in individual bacterial populations was observed within the first month postpartum.
Using partial 16S rRNA sequencing, the vast majority of isolated strains was assigned to the genus Staphylococcus, followed by the strains of Streptococcus and Propionibacterium, and thus showing that facultative anaerobic members of Firmicutes and Actinobacteria predominate in the breast milk ecosystem, while members of Proteobacteria and Bacteroidetes were few or not detected, respectively. Results obtained from the quantification of presumptive populations, however, should be taken with caution due to medium selectivity/electivity bias inherent to culture and the fact that non-fastidious facultative anaerobes are favoured over more fastidious obligate anaerobes, which may thus have escaped culture. For instance, Staphylococcus spp. were able to grow on most culture media, and it can be assumed that they accounted for a high percentage of presumptive total facultative anaerobes and total anaerobes, and consequently that the population of presumptive Streptococcus has been overestimated. However, despite the fact that facultative anaerobes are able to grow in strict anaerobic conditions, the presence of Bifidobacterium as well as Veillonella spp., isolated from the samples of two different mothers each, demonstrates that breast milk can be a source of viable obligate gut-associated anaerobes.
While to our knowledge, no Veillonella spp. have been isolated from breast milk in previous research, all other identified taxa, including the less frequently isolated genera, Rothia, Enterococcus, Escherichia/Shigella and Klebsiella, have been detected in previous studies, although with varying frequencies( Reference Heikkila and Saris 4 , Reference Jimenez, Delgado and Maldonado 6 ). The predominance of Staphylococcus, Streptococcus and Propionibacterium spp. largely confirms the findings of previous culture-dependent studies( Reference Heikkila and Saris 4 – Reference Jimenez, Delgado and Maldonado 6 ). However, in other studies, Enterococcus spp. have also been identified among the dominant populations( Reference Solis, de Los Reyes-Gavilan and Fernandez 5 , Reference Jimenez, Delgado and Maldonado 6 ), while this genus was only identified in two samples in the present study, despite the use of MC agar selective for Enterococcus spp. and Enterobacteriaceae. The presence of Enterococcus spp. in breast milk appears to be largely variable, which is corroborated by quantitative PCR analyses, quantifying this population at 0·5–4 % of the total bacterial DNA( Reference Collado, Delgado and Maldonado 14 , Reference Cabrera-Rubio, Collado and Laitinen 16 ). In contrast to previous research, no viable strains belonging to genera such as Actinomyces and Corynebacterium (Actinobacteria phylum)( Reference Heikkila and Saris 4 , Reference Perez, Dore and Leclerc 8 ), Lactococcus and Leuconostoc (Bacilli class)( Reference Heikkila and Saris 4 ), as well as to the obligate anaerobic genus Peptostreptococcus (Clostridia class)( Reference Perez, Dore and Leclerc 8 ) were isolated by culture; however, these genera were identified as minor components of breast milk microbiota only in these studies.
While culture and 16S rRNA sequencing of the isolates allowed for characterising the major viable bacterial populations in breast milk, pyrosequencing demonstrated that bacterial diversity in breast milk is by far higher than measured by culture-dependent methods, as well as by culture-independent methods with much lower throughput similar to the generation of 16S rRNA clone libraries( Reference Martin, Heilig and Zoetendal 15 ). Taxonomic assignment of the sequences using the Ribosomal Database Project Classifier revealed that, on average, the highest relative abundance was observed for the Firmicutes phylum and, more specifically, the genera Staphylococcus and Streptococcus, which largely supports the findings of our culture-dependent approach. However, in contrast to culture, using pyrosequencing, the relative abundance of Streptococcus was slightly higher than that for Staphylococcus, which may be explained by more fastidious growth requirements of Streptococcus strains.
The second most abundant phylum, Proteobacteria, was largely made up of the genera Pseudomonas and Ralstonia, which is supported by previous culture-independent studies( Reference Martin, Heilig and Zoetendal 15 , Reference Hunt, Foster and Forney 17 ). However, surprisingly, no corresponding strains were detected in our culture-dependent approach, although both MC and nutrient agar are expected to support the growth of such non-fastidious organisms. This suggests that the detected DNA could correspond to stressed, dead or (partially) lysed cells, possibly through the action of antimicrobial components, leucocytes and/or weakness for competition against other bacteria of the breast milk ecosystem. For instance, Lactobacillus spp. isolated from breast milk have been reported as capable of inhibiting nosocomial pathogens, such as strains of Pseudomonas, Escherichia and Serratia ( Reference Jara, Sanchez and Vera 29 ).
In concordance with culture, the Actinobacteria phylum comprised mainly the genus Propionibacterium, and a lower relative abundance of Rothia and Bifidobacterium populations. Hunt et al. ( Reference Hunt, Foster and Forney 17 ) observed a similar bacterial diversity in terms of the fifteen predominant genera in breast milk when using pyrosequencing, but detected only a few sequences of Bifidobacterium, while in the present study, Bifidobacterium was among the twelve most abundant genera. Also, Cabrera-Rubio et al. ( Reference Cabrera-Rubio, Collado and Laitinen 16 ) did not report any pyrosequences belonging assigned to Bifidobacterium. However, the presence of DNA from Bifidobacterium in the present study is supported by previous quantitative PCR analyses quantifying this population in the range of log 2–6 genome equivalents/ml, corresponding to up to 66 % of the total bacterial genome equivalents( Reference Collado, Delgado and Maldonado 14 , Reference Cabrera-Rubio, Collado and Laitinen 16 , Reference Gronlund, Gueimonde and Laitinen 30 ). Besides geographical, dietary, host and sampling time factors, different outcomes in pyrosequencing analyses may be due to differences in methodological factors, such as DNA extraction procedures( Reference Yuan, Cohen and Ravel 31 , Reference Maukonen, Simoes and Saarela 32 ), PCR bias and different taxonomic classification accuracies due to the use of primers targeted at different tandem variable 16S rRNA gene regions( Reference Claesson, Wang and O'Sullivan 33 , Reference Wu, Lewis and Hoffmann 34 ) (i.e. the V1–V2 region in both previous studies( Reference Cabrera-Rubio, Collado and Laitinen 16 , Reference Hunt, Foster and Forney 17 ), compared with the V5–V6 region in the present study), as well as a higher depth of sequencing in the present study (i.e. average 5051 reads/sample) compared with both previous studies (i.e. average 2623( Reference Cabrera-Rubio, Collado and Laitinen 16 ) and 3400 reads/sample( Reference Hunt, Foster and Forney 17 )). This may also explain further differences, such as the higher relative abundance of Corynebacterium and Serratia in some of the samples analysed by Hunt et al. ( Reference Hunt, Foster and Forney 17 ), as well as the predominance of Weissella and Leuconostoc over Staphylococcus and Streptococcus in the study by Cabrera-Rubio et al. ( Reference Cabrera-Rubio, Collado and Laitinen 16 ). Furthermore, a higher relative abundance of the anaerobic genera Prevotella and Veillonella was observed in both previous pyrosequencing studies( Reference Cabrera-Rubio, Collado and Laitinen 16 , Reference Hunt, Foster and Forney 17 ); however, this may be due to the fact that samples were collected at a later stage (i.e. 1–10 months postpartum), when compared with within the first month postpartum in the present study.
Much focus has been given to the presence of the potential probiotic Lactobacillus spp. in breast milk( Reference Solis, de Los Reyes-Gavilan and Fernandez 5 , Reference Albesharat, Ehrmann and Korakli 7 , Reference Jara, Sanchez and Vera 29 , Reference Martin, Langa and Reviriego 35 , Reference Martin, Jimenez and Olivares 36 ). However, the mean relative abundance of Lactobacillus was generally low in the present study group (i.e. < 0·1 %), which is in accordance with the low isolation frequency of Lactobacillus spp. (i.e. L. gasseri and L. brevis) observed using culture. Therefore, the supplementation of formulae with probiotic strains of Lactobacillus may not reflect the natural bacterial inoculum transferred to the breast-fed neonate, despite their potential for disease prevention and treatment( Reference Thomas and Greer 37 ). Differences in bacterial densities may, however, be attributed to geographical factors, including dietary habits, such as the consumption of probiotic products. In this regard, it has been shown that Lactobacillus spp. when administered orally can be recovered from breast milk( Reference Jimenez, Fernandez and Maldonado 38 – Reference Abrahamsson, Sinkiewicz and Jakobsson 40 ) and thus influence its composition.
Pyrosequencing largely confirmed the culture-dependent approach by the detection of Staphylococcus, Streptococcus and Propionibacterium spp. as dominant populations, which as facultative anaerobic skin/oral-associated populations may be transferred to the breast by secondary contamination such as via suckling. Interestingly, however, pyrosequencing also allowed for detecting a series of obligate anaerobic, gut-associated genera, such as Bacteroides and multiple members of the Clostridia class, in addition to Bifidobacterium and Veillonella also identified by culture. Among the members of the Clostridia, genera such as Blautia, Dorea and Ruminococcus were detected for the first time and, moreover, surprisingly, also major butyrate producers important for colonic health, such as Faecalibacterium, Roseburia, Coprococcus and Subdoligranulum. The present findings are supported by and extend previous quantitative PCR analyses confirming, although at a higher taxonomic level only, the presence of the Bacteroides group, the Clostridium coccoides group, as well as the Clostridium clusters IV and XIV that comprise the major gut-associated butyrate producers, Faecalibacterium and Roseburia, respectively( Reference Collado, Delgado and Maldonado 14 , Reference Cabrera-Rubio, Collado and Laitinen 16 ). Thus, breast milk may represent a source of such obligate anaerobes to the breast-fed neonate and may contribute to the colonisation process, as suggested in our previous research( Reference Jost, Lacroix and Braegger 13 ) by the presence of Bifidobacterium, Bacteroides and Veillonella spp. as pioneer bacteria in corresponding neonatal faeces. We hypothesise that Veillonella spp., as non-carbohydrate fermenting, lactate utilisers (in concert with Propionibacterium spp.), may play an important function in the breast milk ecosystem by metabolising lactate resulting from lactose fermentation by predominant breast milk populations, to propionate and acetate( Reference Duncan, Louis and Flint 41 ). These trophic functions may be transferred to the breast-fed neonate and contribute to the colonisation and gut maturation process. Furthermore, the detection of the members of Bacteroidetes and Clostridia in breast milk suggests that they originate from the maternal gut, since such obligate anaerobes do not proliferate outside their host. This hypothesis, i.e. the translocation of maternal bacteria to subsequently reach the mammary gland via a bacterial enteromammary pathway, has been proposed previously( Reference Perez, Dore and Leclerc 8 , Reference Martín, Langa and Reviriego 42 ) and will be the object of future research.
Furthermore, it remains to be elucidated whether the series of anaerobes detected using pyrosequencing escaped culture due to their low abundance in breast milk and/or due to fastidious growth requirements (viable but not culturable with the applied methods), and/or the presence of antimicrobial compounds, or if dead cells or parts thereof are transferred to the breast-fed neonate, which nevertheless could elicit an immune response and affect neonatal development. Although, in the present study, samples were processed under anaerobic conditions using culture media supporting the growth of gut-associated anaerobes, a higher number of isolates may be necessary for identifying subdominant populations. On the other hand, highly fastidious anaerobes may be recovered using more laborious roll-tube techniques developed by Hungate( 43 ) and approaches like the recently described ‘microbial culturomics’( Reference Lagier, Armougom and Million 44 ) that rely on a vast array of culture conditions.
Acknowledgements
The present study was supported by Nestlé Nutrition (Vevey, Switzerland), Nestec (Lausanne, Switzerland) and the Swiss Foundation for Nutrition Research (SFEFS) (Zurich, Switzerland) for the purpose of basic research only and is not related to any patent, product in development or marketed product. None of the authors are employees or consultants of the funders. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors are grateful to Patrick Bühr, Michael Friedt, Petra Martel, Daniela Rogler and Rebekka Koller at the University Children's Hospital (Zurich, Switzerland) for their effort in volunteer recruitment and sampling, to Peter Frei and Tania Torossi for their assistance in 16S rRNA gene sequencing, carried out at the Genetic Diversity Centre of ETH Zurich (Zurich, Switzerland), as well as to Medela AG (Baar, Switzerland) for providing electrical breast pumps throughout the study.
C. B., C. L., C. C. and T. J. conceived and designed the experiments. T. J. and C. C. acquired the data. T. J., C. C., C. L. and C. B. analysed and interpreted the data. T. J., C. C., C. L. and C. B. wrote and approved the manuscript. The authors declare that no competing interests exist.





