In this issue of the British Journal of Nutrition, Dr Jean-Philippe Bonjour presents a very well-organised and comprehensive review of the literature on how acids and bases are produced in the body(Reference Bonjour1). The review explains how the body buffers and titrates acids and bases, and discusses whether dietary factors can affect systemic acid–base status and balance and whether dietary acid factors can affect the bone sufficiently to cause bone breakdown leading to osteoporosis. Osteoporosis is considered to be a state where bone breakdown exceeds bone production, leading to both the progressive loss of bone mass and the destruction of bone architecture. As noted by Dr Bonjour, the idea that acidosis-induced bone mass reduction can occur and that alkali administration might slow or prevent this cause of bone loss was first proposed more than 40 years ago. Over the past two decades, opposition to the idea that alkali therapy may help prevent dietary acid-induced osteoporosis has been growing, as studies that failed to show support for this theory were published. Presently, data that support both the proponents and the opponents of this hypothesis exist.
Proponents of the theory argue that in vitro studies clearly show that pronounced increases in systemic acid levels activate osteoclasts and cause increased flux of Ca from bone and increased bone dissolution(Reference Barzel2–Reference Frick, Krieger and Nehrke6). Therefore, smaller changes towards a higher blood and tissue acid steady-state level associated with typical net acid-producing diets, exacerbated by age-related decline in renal acid–base regulatory ability, may cause smaller changes that over decades become clinically significant. This effect would exacerbate the bone mass-reducing effect of known aetiological factors contributing to osteoporosis. They then cite studies that support this hypothesis(Reference Sellmeyer, Stone and Sebastian7–Reference Jehle, Hulter and Krapf11).
Opponents of this theory argue from quantitative considerations that if bone were the main site of deposition of the base used to titrate dietary acids, then all the bone in the body would be dissolved in just a few years. They point out that because that does not occur, bone cannot provide the major quantity of base (alkali) titrating towards the neutralisation of dietary net acid(Reference Oh12–Reference Oh14). They then cite supporting studies(Reference Macdonald, Black and Aucott15–Reference Mardon, Habauzit and Trzeciakiewicz19). Dr Bonjour(Reference Bonjour1) also argues that homeostatic mechanisms, including renal net acid excretion (NAE), would not permit a steady-state low-grade metabolic acidosis caused by the ingestion of typical Western net acid-producing diets.
Is it possible to reconcile these two disparate points of view? We suggest that both points of view are partially correct and offer further suggestions to integrate these hypotheses.
Do higher dietary acid loads, in fact, lead to higher steady-state blood acid levels? Kurtz et al. (Reference Kurtz, Maher and Hulter20) and Frassetto et al. (Reference Frassetto, Morris and Sebastian21) showed that in healthy humans, consuming ordinary diets, the steady-state blood hydrogen ion concentration is detectably higher, and the plasma bicarbonate concentration detectably lower when dietary net acid loads are higher, within the range typically observed in American and European diets. Homeostatic mechanisms did not maintain hydrogen ion or bicarbonate levels when the diets yielded acid loads greater than approximately 1 mmol/kg per d and net acid balance became positive (see Fig. 1). However, the typical American or Western diet averages approximately 50 mmol/d(Reference Lemann22), within the range where endogenous acid production (including dietary acid intake) is matched by renal NAE in subjects with normal renal function. In these people, the kidneys are able to excrete the entire acid load, as suggested by Dr Bonjour(Reference Bonjour1).
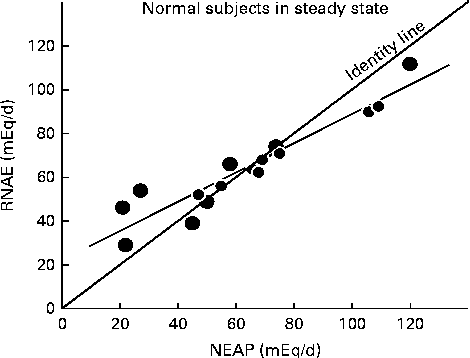
Fig. 1 Relationship between steady-state renal net acid excretion (RNAE) and net endogenous acid production (NEAP) in normal subjects ingesting one of three different diets(Reference Sebastian, Frassetto, Merriam, Gennari, Adrogue, Galla and Madias29). Each data point represents the mean steady-state value observed in one individual (r 0·94, P< 0·01). Note that as NEAP increases, RNAE falls below the ‘identity’ line, where NEAP = RNAE, suggesting that at higher acid loads, the kidneys are not able to excrete all of the increased acids. This then produces a positive net acid balance, reflected by the lower blood pH and serum bicarbonate values demonstrated by Kurtz et al. (Reference Kurtz, Maher and Hulter20) and Frassetto et al. (Reference Frassetto, Morris and Sebastian21) in subjects with high dietary acid loads. Also note that in subjects on low endogenous acid diets, RNAE exceeds the identity line, as predicted by Hood & Tannen(Reference Hood and Tannen24). Data are plotted from tabular data published in Lennon et al. (Reference Lennon, Lemann and Litzow30).
Do other osteoporosis risk factors have a larger impact on bone? If many factors of differing magnitude of effect by themselves and/or in synergistic interactions among them contribute to the development of osteoporotic low bone mass, and if many different partially counteracting homeostatic mechanisms come into play, then it might be difficult to detect and quantify a negative effect of habitual consumption of typical net acid-producing diets, in particular if such a negative effect is quantitatively relatively small. For example, age, sex, weight and immobility are thought to have more relative impact on fracture risk than coffee drinking, smoking or trace mineral levels(Reference Sellmeyer, Stone and Sebastian7, 23); diet acid load may be another quantitatively small factor.
Finally, assuming that bone is not the predominate system for neutralising acid loads, what other systems could contribute to maintaining net acid balance? Other potential systems for buffering or titrating acids have been demonstrated. Titration is used in this sense as a system that does not regenerate itself, when compared with buffering, which does.
Hood & Tannen(Reference Hood and Tannen24) suggested in 1998 that systemic pH was protected by increasing or decreasing organic acid production in the direction that attenuates the change in systemic pH. In overweight humans fasting or placed on ketogenic diets, addition of ammonium chloride, an acid, caused decreases in urinary ketoacid excretion compared with the controls on NaCl, while those given sodium bicarbonate exhibited increased urinary ketoacid excretion. These studies suggest that alteration in organic acid production is one of the main methods that the body uses to maintain systemic blood acid levels.
Wesson et al. (Reference Wesson, Simoni and Broglio25) suggested that tissue buffering by the kidney was important, and showed that chronic alkali loading in subjects with chronic kidney disease stage 1 (glomerular filtration rate (GFR) >90 ml/min per 1·73 m2 and with proteinuria) and stage 2 (GFR 60–90 ml/min per 1·73 m2) caused no change in blood pH or bicarbonate, but in the stage 2 chronic kidney disease (CKD) subjects allowed the kidneys to lower NAE by 30 %(Reference Mahajan, Simoni and Sheather26). This level of renal insufficiency is that seen in older subjects with age-related decline in kidney function. In this study, alkali therapy slowed the progression of GFR decline in subjects with CKD stage 2, but not in subjects with CKD stage 2 on NaCl or placebo.
Muscle is another tissue that responds to high acid loads, buffering hydrogen ions intracellularly by exchanging them for K and by titration through proteolysis. High acid environments up-regulate the ubiquitin–proteasome pathway, leading to increased glutamine available for the kidney(Reference Mitch, Medina and Grieber27). The kidney uses glutamine to increase urinary ammonium excretion, which mitigates the increased acid load. Alkali therapy may alleviate some of that muscle breakdown and possibly improve muscle function (reviewed in Mithal et al. (Reference Mithal, Bonjour and Boonen28)).
Thus, given all the other factors that could also have an impact on the development of osteoporosis and the body's systems for buffering and titrating systemic acid levels, people with relatively normal kidneys, eating typical Western diets with acid loads up to 1 mmol/kg, will not show net retention of a portion of their net endogenous acid production and therefore would not be candidates for a bone-ameliorative effect of neutralising their dietary net endogenous acid load. This suggests that subjects who are elderly with lower renal function and low muscle and bone mass are most at risk if they eat diets with high acid loads and would be the group whose bones (and muscles and kidneys!) would most demonstrably benefit from alkali therapies.




In this issue of the British Journal of Nutrition, Dr Jean-Philippe Bonjour presents a very well-organised and comprehensive review of the literature on how acids and bases are produced in the body(Reference Bonjour1). The review explains how the body buffers and titrates acids and bases, and discusses whether dietary factors can affect systemic acid–base status and balance and whether dietary acid factors can affect the bone sufficiently to cause bone breakdown leading to osteoporosis. Osteoporosis is considered to be a state where bone breakdown exceeds bone production, leading to both the progressive loss of bone mass and the destruction of bone architecture. As noted by Dr Bonjour, the idea that acidosis-induced bone mass reduction can occur and that alkali administration might slow or prevent this cause of bone loss was first proposed more than 40 years ago. Over the past two decades, opposition to the idea that alkali therapy may help prevent dietary acid-induced osteoporosis has been growing, as studies that failed to show support for this theory were published. Presently, data that support both the proponents and the opponents of this hypothesis exist.
Proponents of the theory argue that in vitro studies clearly show that pronounced increases in systemic acid levels activate osteoclasts and cause increased flux of Ca from bone and increased bone dissolution(Reference Barzel2–Reference Frick, Krieger and Nehrke6). Therefore, smaller changes towards a higher blood and tissue acid steady-state level associated with typical net acid-producing diets, exacerbated by age-related decline in renal acid–base regulatory ability, may cause smaller changes that over decades become clinically significant. This effect would exacerbate the bone mass-reducing effect of known aetiological factors contributing to osteoporosis. They then cite studies that support this hypothesis(Reference Sellmeyer, Stone and Sebastian7–Reference Jehle, Hulter and Krapf11).
Opponents of this theory argue from quantitative considerations that if bone were the main site of deposition of the base used to titrate dietary acids, then all the bone in the body would be dissolved in just a few years. They point out that because that does not occur, bone cannot provide the major quantity of base (alkali) titrating towards the neutralisation of dietary net acid(Reference Oh12–Reference Oh14). They then cite supporting studies(Reference Macdonald, Black and Aucott15–Reference Mardon, Habauzit and Trzeciakiewicz19). Dr Bonjour(Reference Bonjour1) also argues that homeostatic mechanisms, including renal net acid excretion (NAE), would not permit a steady-state low-grade metabolic acidosis caused by the ingestion of typical Western net acid-producing diets.
Is it possible to reconcile these two disparate points of view? We suggest that both points of view are partially correct and offer further suggestions to integrate these hypotheses.
Do higher dietary acid loads, in fact, lead to higher steady-state blood acid levels? Kurtz et al. (Reference Kurtz, Maher and Hulter20) and Frassetto et al. (Reference Frassetto, Morris and Sebastian21) showed that in healthy humans, consuming ordinary diets, the steady-state blood hydrogen ion concentration is detectably higher, and the plasma bicarbonate concentration detectably lower when dietary net acid loads are higher, within the range typically observed in American and European diets. Homeostatic mechanisms did not maintain hydrogen ion or bicarbonate levels when the diets yielded acid loads greater than approximately 1 mmol/kg per d and net acid balance became positive (see Fig. 1). However, the typical American or Western diet averages approximately 50 mmol/d(Reference Lemann22), within the range where endogenous acid production (including dietary acid intake) is matched by renal NAE in subjects with normal renal function. In these people, the kidneys are able to excrete the entire acid load, as suggested by Dr Bonjour(Reference Bonjour1).
Fig. 1 Relationship between steady-state renal net acid excretion (RNAE) and net endogenous acid production (NEAP) in normal subjects ingesting one of three different diets(Reference Sebastian, Frassetto, Merriam, Gennari, Adrogue, Galla and Madias29). Each data point represents the mean steady-state value observed in one individual (r 0·94, P< 0·01). Note that as NEAP increases, RNAE falls below the ‘identity’ line, where NEAP = RNAE, suggesting that at higher acid loads, the kidneys are not able to excrete all of the increased acids. This then produces a positive net acid balance, reflected by the lower blood pH and serum bicarbonate values demonstrated by Kurtz et al. (Reference Kurtz, Maher and Hulter20) and Frassetto et al. (Reference Frassetto, Morris and Sebastian21) in subjects with high dietary acid loads. Also note that in subjects on low endogenous acid diets, RNAE exceeds the identity line, as predicted by Hood & Tannen(Reference Hood and Tannen24). Data are plotted from tabular data published in Lennon et al. (Reference Lennon, Lemann and Litzow30).
Do other osteoporosis risk factors have a larger impact on bone? If many factors of differing magnitude of effect by themselves and/or in synergistic interactions among them contribute to the development of osteoporotic low bone mass, and if many different partially counteracting homeostatic mechanisms come into play, then it might be difficult to detect and quantify a negative effect of habitual consumption of typical net acid-producing diets, in particular if such a negative effect is quantitatively relatively small. For example, age, sex, weight and immobility are thought to have more relative impact on fracture risk than coffee drinking, smoking or trace mineral levels(Reference Sellmeyer, Stone and Sebastian7, 23); diet acid load may be another quantitatively small factor.
Finally, assuming that bone is not the predominate system for neutralising acid loads, what other systems could contribute to maintaining net acid balance? Other potential systems for buffering or titrating acids have been demonstrated. Titration is used in this sense as a system that does not regenerate itself, when compared with buffering, which does.
Hood & Tannen(Reference Hood and Tannen24) suggested in 1998 that systemic pH was protected by increasing or decreasing organic acid production in the direction that attenuates the change in systemic pH. In overweight humans fasting or placed on ketogenic diets, addition of ammonium chloride, an acid, caused decreases in urinary ketoacid excretion compared with the controls on NaCl, while those given sodium bicarbonate exhibited increased urinary ketoacid excretion. These studies suggest that alteration in organic acid production is one of the main methods that the body uses to maintain systemic blood acid levels.
Wesson et al. (Reference Wesson, Simoni and Broglio25) suggested that tissue buffering by the kidney was important, and showed that chronic alkali loading in subjects with chronic kidney disease stage 1 (glomerular filtration rate (GFR) >90 ml/min per 1·73 m2 and with proteinuria) and stage 2 (GFR 60–90 ml/min per 1·73 m2) caused no change in blood pH or bicarbonate, but in the stage 2 chronic kidney disease (CKD) subjects allowed the kidneys to lower NAE by 30 %(Reference Mahajan, Simoni and Sheather26). This level of renal insufficiency is that seen in older subjects with age-related decline in kidney function. In this study, alkali therapy slowed the progression of GFR decline in subjects with CKD stage 2, but not in subjects with CKD stage 2 on NaCl or placebo.
Muscle is another tissue that responds to high acid loads, buffering hydrogen ions intracellularly by exchanging them for K and by titration through proteolysis. High acid environments up-regulate the ubiquitin–proteasome pathway, leading to increased glutamine available for the kidney(Reference Mitch, Medina and Grieber27). The kidney uses glutamine to increase urinary ammonium excretion, which mitigates the increased acid load. Alkali therapy may alleviate some of that muscle breakdown and possibly improve muscle function (reviewed in Mithal et al. (Reference Mithal, Bonjour and Boonen28)).
Thus, given all the other factors that could also have an impact on the development of osteoporosis and the body's systems for buffering and titrating systemic acid levels, people with relatively normal kidneys, eating typical Western diets with acid loads up to 1 mmol/kg, will not show net retention of a portion of their net endogenous acid production and therefore would not be candidates for a bone-ameliorative effect of neutralising their dietary net endogenous acid load. This suggests that subjects who are elderly with lower renal function and low muscle and bone mass are most at risk if they eat diets with high acid loads and would be the group whose bones (and muscles and kidneys!) would most demonstrably benefit from alkali therapies.
Acknowledgements
The authors state that they have no conflict of interest.