


Mitochondria are often referred to as the powerhouse of the cell because they consume nearly 90 % of cells' oxygen to support oxidative phosphorylation for the synthesis of ATP, the energy currency used for a variety of metabolic reactions. Mitochondrial ATP production occurs when electrons are passed along a series of molecular complexes in the inner mitochondrial membrane known as the electron transport chain. The energy released by the flow of electrons through the electron transport chain is used to transfer protons across the inner mitochondrial membrane, creating a large mitochondrial membrane potential, which is used to drive ATP synthesis as protons re-enter the mitochondrial matrix through the F0F1 ATP synthase(Reference Stock, Gibbons and Arechaga1). Ideally, the transfer of electrons across the electron transport chain should result in the four-electron reduction of molecular oxygen (O2) to water (H2O). However, this is not always the case.
During the transfer of electrons along the electron transport chain, single electrons sometimes escape and result in a single electron reduction of molecular oxygen to form a superoxide anion (![]() )(Reference Fariss, Chan and Patel2). It is estimated that as much as 1 % of all oxygen consumed may result in the formation of reactive oxygen species (ROS) such as
)(Reference Fariss, Chan and Patel2). It is estimated that as much as 1 % of all oxygen consumed may result in the formation of reactive oxygen species (ROS) such as ![]() , with the vast majority of ROS being generated in the mitochondria(Reference Ischiropoulos and Beckman3), supporting the prevailing opinion that the mitochondria are the major site of ROS production in the cell and therefore the prime targets for oxidative damage(Reference Trifunovic and Larsson4). Superoxide may spontaneously dismute, or the dismutation may be catalysed by superoxide dismutase (SOD), forming H2O2. This can cause oxidative damage to the surrounding biomolecules, as, in the presence of ferrous Fe, H2O2 may react to form the highly reactive hydroxyl radical, via Fenton chemistry.
, with the vast majority of ROS being generated in the mitochondria(Reference Ischiropoulos and Beckman3), supporting the prevailing opinion that the mitochondria are the major site of ROS production in the cell and therefore the prime targets for oxidative damage(Reference Trifunovic and Larsson4). Superoxide may spontaneously dismute, or the dismutation may be catalysed by superoxide dismutase (SOD), forming H2O2. This can cause oxidative damage to the surrounding biomolecules, as, in the presence of ferrous Fe, H2O2 may react to form the highly reactive hydroxyl radical, via Fenton chemistry.
In order to combat the effects of ROS, aerobic organisms have developed an extensive antioxidant defence system, which include both non-enzymatic and enzymatic antioxidants. Enzymatic antioxidants include SOD, glutathione peroxidase (GPx) and catalase(Reference Mates, Perez-Gomez and Nunez de Castro5), whereas non-enzymatic antioxidants are represented by vitamin C, vitamin E, thiol antioxidants (glutathione and lipoic acid) and other antioxidants(Reference McCall and Frei6). However, when these ROS are chronically produced at high rates, they may overwhelm non-enzymatic and enzymatic antioxidant systems. As a result, ROS could then cause deleterious effects by inducing the irreversible oxidation of their principal targets such as DNA, lipids and proteins(Reference Valko, Leibfritz and Moncol7), leading to oxidative damage and cell death.
Oxidative damage to the mitochondria has been proposed to cause a wide range of metabolic disorders including Parkinson's disease(Reference Henchcliffe and Beal8, Reference Forno9), Huntington's disease(Reference Squitieri, Cannella and Sgarbi10), Alzheimer's disease(Reference Mattson and Magnus11, Reference Beal12), diabetes(Reference Evans, Goldfine and Maddux13), obesity(Reference Wlodek and Gonzales14), fatty liver(Reference Song, Moon and Olsson15) and ageing(Reference Stadtman16, Reference Harman17). For the last three decades, antioxidant therapy has been intensively studied to prevent or treat these oxidative stress-associated diseases. Although antioxidants such as vitamin E, ubiquinol and N-acetylcysteine have previously been reported to decrease mitochondrial ROS concentrations and consequently alleviate mitochondrial oxidative damage(Reference Matthews, Yang and Browne18–Reference Sokol, McKim and Goff20), their effectiveness still seems to be largely limited by their poor capacity to accumulate specifically within the mitochondria.
Recently, Murphy and co-workers(Reference Smith, Porteous and Gane21, Reference Murphy and Smith22) developed a series of mitochondria-targeted antioxidants by conjugation with triphenylphosphonium cations, which can permeate lipid bilayers easily and accumulate 100- to 1000-fold within the mitochondria. One of these mitochondria-targeted antioxidants is a mitochondria-targeted vitamin E derivative (MitoVit E), which has been tested in a wide range of mitochondrial and cell models. Indeed, it has been shown to mitigate ethanol-induced suppression of antioxidant defence systems in cerebellar granule cells(Reference Siler-Marsiglio, Pan and Paiva23), inhibit peroxide-induced apoptosis in endothelial cells(Reference Dhanasekaran, Kotamraju and Kalivendi24) and prevent cell death in Friedreich's ataxia fibroblasts(Reference Jauslin, Meier and Smith25). However, to our knowledge, the effect of MitoVit E on mitochondrial alterations and systemic oxidative stress has never been studied in vivo. In order to investigate this further, we investigated the effects of MitoVit E on mitochondrial ATP and H2O2 production rates, mitochondrial size and number, urinary isoprostane concentration, and some plasma redox biomarkers in C57BL/6 mice.
Materials and methods
Chemicals
MitoVit E was synthesised with the starting material benzopyran-6-ol acetate, which was prepared in one step from commercially available chemicals as reported by Ichikawa & Kato(Reference Ichikawa and Kato26). Benzopyran-6-ol acetate was oxidised using ozone in methylene chloride–methanol at − 78°C. The resulting solution was purged with Ar to remove excess ozone and then treated directly with sodium borohydride to produce an alcohol similar to that in the scientific literature(Reference Ichikawa and Kato26, Reference Spivak, Knyshenko and Mallyabaeva27). The alcohol was converted to MitoVit E as reported by Grisar et al. (Reference Grisar, Petty and Bolkenius28). Briefly, the alcohol was treated with iodine, triphenylphosphine and imidazole in methylene chloride at 0°C for 3 h to obtain a 92 % iodide yield. The iodide was treated with triphenylphosphine in acetonitrile at reflux for 11 h to produce the phosphonium salt as a crystalline solid in quantitative yield. The product of MitoVit E was >95 % pure as evidenced by TLC and NMR.
All the other chemicals used in the present study were obtained from Sigma-Aldrich (St Louis, MO, USA), unless otherwise stated.
Animals and study design
A total of sixty-four 7-week-old C57BL/6 male mice (Jackson Laboratory, Bar Harbor, ME, USA) were divided into four blocks and arrived at four different times. Mice were housed one per cage in a temperature-controlled (18–22°C) and light-controlled environment with a 12 h light–12 h dark cycle. Mice were fed a high-fat (HF) diet for 5 weeks and then switched to either a low-fat (LF) diet or a medium-fat (MF) diet, and administered orally with MitoVit E (40 mg MitoVit E/kg body weight) or drug vehicle (10 % (v/v) ethanol in 0·9 % (w/v) NaCl solution), every other day for 5 weeks. Detailed compositions of the HF, MF and LF diets are provided in Table S1 of the supplementary material (available online at http://www.journals.cambridge.org/bjn). All mice in the four groups (LF group receiving drug vehicle, LF-C; LF group receiving MitoVit E, LF-E; MF group receiving drug vehicle, MF-C; MF group receiving MitoVit E, MF-E) had unrestricted access to feed and water. During the study, urine was collected for isoprostane and creatinine assays. At the end of the study, all mice were killed by exposure to CO2 in a gas chamber. Blood was collected by cardiac puncture for biochemical assays in the plasma. The gastrocnemius and liver from eight mice of each group were rapidly excised for mitochondrial isolation and subsequent measurements of ATP and H2O2 production rates in the mitochondria. The soleus muscles from two mice of each group were used in transmission electron microscopy for quantification of mitochondrial number and average size. Institutional and national guidelines for the care and use of animals were followed, and all experimental procedures involving animals were approved by the Institutional Animal Care and Use Committee of Iowa State University.
Mitochondrial preparation from the liver and skeletal muscle
Liver mitochondria were isolated at 4°C, as described previously by Cawthon et al. (Reference Cawthon, McNew and Beers29), with some modifications. Briefly, liver tissue (0·5 g) was homogenised using a Potter–Elvehjem glass homogeniser (Omni International, Kennesaw, GA, USA) in 10 ml iced extraction buffer (10 mm-HEPES, 200 mm-mannitol, 70 mm-sucrose, 1 mm-ethylene glycol tetra-acetic acid and bovine serum albumin (2 g/l), pH 7·5). Homogenates were centrifuged at 600 g for 8 min, and the supernatants were filtered through a double layer of cheesecloth followed by centrifugation at 11 000 g for 12 min. The pellet was resuspended in 20 ml extraction buffer without bovine serum albumin and centrifuged at 600 g for 8 min. The supernatants were centrifuged at 11 000 g for 12 min to get the final mitochondrial pellets for subsequent analyses.
Skeletal muscle mitochondria were isolated at 4°C, as described previously by Chappell & Perry(Reference Chappell and Perry30), with slight modifications. The gastrocnemius was minced with scissors in Chappell–Perry Medium I (100 mm-KCl, 50 mm-Tris–HCl, 5 mm-MgCl2, 1 mm-EDTA and 1 mm-ATP, pH 7·2) and incubated in 20 vol. of Chappell–Perry Medium I supplemented with Nagarse (150 mg/l; Sigma-Aldrich) for 10 min. The muscles were then homogenised at 4°C with a Potter–Elvehjem glass homogeniser (Omni International). After the homogenate was centrifuged at 600 g for 10 min, the supernatant was filtered through a double layer of cheesecloth and centrifuged at 14 000 g for 10 min. The pellets were resuspended in modified Chappell–Perry II buffer (100 mm-KCl, 50 mm-Tris–HCl, 1 mm-MgCl2, 0·2 mm-EDTA and 1 mm-ATP, pH 7·2) with 0·5 % (w/v) bovine serum albumin and centrifuged at 7000 g for 10 min. The pellets were resuspended in modified Chappell–Perry II buffer without bovine serum albumin and centrifuged at 3500 g for 10 min; this step was repeated twice. Mitochondrial pellets were used for subsequent analyses.
Determination of ATP and hydrogen peroxide production rates in the mitochondria
ATP production rate was measured, as described previously by Mansouri et al. (Reference Mansouri, Muller and Liu31). Reactions were conducted in ninety-six-well microplates with a 100 μl reaction system (25 mm-Tricine, 5 mm-MgSO4, 0·1 mm-EDTA and 0·1 mm-NaN3, pH 7·8) containing 100 μg of mitochondrial protein, substrates (1 mm-glutamate plus 1 mm-malate), ADP (0·025 mmol/l) and luciferase reagent (Roche, Madison, WI, USA). Luminescence was recorded by the Synergy™ 2 Multi-Mode microplate reader (BioTek Instruments, Winooski, VT, USA) every 36 s for a total of 6 min. The slope of increase within the linear portion of the curve was used for the ATP production rate.
Mitochondrial H2O2 production rate was measured by using Amplex™ Red–horseradish peroxidase (N-acetyl-3,7-dihydroxyphenoxazine; Invitrogen, Carlsbad, CA, USA), as described previously by Muller et al. (Reference Muller, Liu and Van Remmen32). Sample reactions were conducted in ninety-six-well microplates with a 100 μl reaction system (125 mm-KCl, 10 mm-HEPES, 2 mm-K2HPO4 and 5 mm-MgCl2) containing 40 μg of mitochondrial protein, substrate (9 mm-succinate), CuZnSOD (100 000 units/l), Amplex™ Red reagent (80 μmol/l) and horseradish peroxidase (1000 units/l). Fluorescence was recorded at an excitation wavelength of 530 nm and an emission wavelength of 590 nm on the Synergy™ 2 Multi-Mode microplate reader (BioTek Instruments) every 36 s for a total of 10 min. The slope of increase within the linear portion of the curve was used for the H2O2 production rate.
For the two aforementioned assays, sample blanks contained all components in the reaction system except mitochondrial protein. The slopes of the increase in the blanks were subtracted from the slopes of the increase in the samples. The results for ATP and H2O2 production rates were analysed by Gen5 (BioTek Instruments).
Measurement of mitochondrial size and number
Soleus muscle was excised from mice and immediately immersed in a cold (4°C) primary fixative consisting of 2 % (w/v) glutaraldehyde and 2 % (w/v) paraformaldehyde in a 0·1 m-cacodylate buffer at pH 7·24. The muscle was sliced into smaller pieces in the fixative and stored overnight at 4°C. The pieces were washed in cold buffer (three times, 20 min each) and placed into 1 % (w/v) OsO4 in the same buffer for 1 h. After this secondary fixation, the pieces were washed several times in the buffer, placed into 2 % (w/v) aqueous uranyl acetate for 1 h and dehydrated through an ethanol series to pure ethanol and ultra-pure ethanol before infiltration with increasing concentrations of Embed 812 resin mixture to pure resin mixture. Pieces were embedded in moulds and polymerised at 60°C for 24 h. The pieces were sectioned (1 μm thick) with glass knives using a Leica–Reichert ultramicrotome (www.leica.com) to determine orientation and location. Thin sections (60–90 nm) thick were cut with a diamond knife and placed on 200 mesh Cu grids. Images were made using a JEOL 2100 TEM (www.jeol.com) set at a magnification (4000 × ) with a bar scale on each image. The images were transferred into an SIS Analysis (Olympus, Münster, Germany; www.soft-imaging.net) program that calculated mitochondrial number and individual area.
Measurement of urinary isoprostane and creatinine
Urine samples were thawed at room temperature and vortexed vigorously before analysis. Urinary 8-iso-PGF2α was determined using a commercially available ELISA kit from Oxford Biomedical Research (Oxford, MI, USA). Internal controls were run on every plate to adjust the plate differences. Urinary creatinine was quantified with a commercial kit from Cayman Chemical (Ann Arbor, MI, USA) using a method based on the reaction of creatinine and alkaline picrate according to the manufacturer's instructions. Results of urinary isoprostane were normalised by urinary creatinine.
Measurement of redox parameters in the plasma
Activities of plasma SOD and GPx were measured using kits from Cayman Chemical. Plasma H2O2 concentration was measured using an Amplex Red hydrogen peroxide assay kit from Invitrogen.
Determination of protein concentration
Protein concentration was measured using the Bradford method(Reference Bradford33).
Statistical analysis
Data were analysed by ANOVA using a general randomised complete block design, with each of the four different arrival times considered as a block. Block, diet, MitoVit E and their interaction were the fixed effects in the model. SAS Proc Mixed (SAS, 2005; SAS Institute, Cary, NC, USA) was used to compute the ANOVA table, least-squares means and differences between the least-squares means. A block effect representing differences between the four blocks could be attributed to many factors (e.g. four blocks of mice were different, or assay reagents or set-ups in the four individual experiments were different). The block effect is not discussed in the present study, but it has to be ruled out for correctly evaluating the effects of the MF diet and MitoVit E. Data are presented as means with their standard errors. Differences between groups were considered to be significant at P < 0·05.
Results
Levels of urinary isoprostane and mouse body weight before treatments
Isoprostanes are a unique series of PG-like compounds formed in vivo from the free radical-catalysed peroxidation of arachidonic acid independent of cyclo-oxygenase enzymes(Reference Morrow and Roberts34). The quantification of plasma or urinary F2-isoprostanes has been regarded as one of the most accurate methods to assess in vivo oxidant stress status when compared with other biomarkers(Reference Kadiiska, Gladen and Baird35). Levels of urinary isoprostane increased 6- and 7·5-fold, respectively, after 2 and 4 weeks of HF feeding before either drug vehicle or MitoVit E administration (Fig. 1(a)). Body weight also increased significantly (P < 0·05) during this period (Fig. 1(b)).
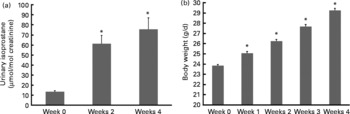
Fig. 1 (a) Urinary isoprostane concentration of mice in the first 4 weeks when fed a high-fat (HF) diet (n 48). Urinary isoprostane was normalised by urinary creatinine (μmol/mol creatinine). (b) Body weight of mice in the first 4 weeks when fed a HF diet (g/d, n 64). Values are means, with standard errors represented by vertical bars. * Mean values were significantly different from those of week 0 (P < 0·05).
Mitochondrial ATP and hydrogen peroxide production rates
In both the liver and muscle, ATP and H2O2 production rates were not affected by either MitoVit E administration or the MF diet when succinate was used as the substrate (Table 1). ATP production rate in the muscle mitochondria was almost as twentyfold high as that in the liver mitochondria, whereas H2O2 production rate in the liver mitochondria was as tenfold high as that in the muscle mitochondria (Table 1).
Table 1 ATP and hydrogen peroxide production rates in the liver and muscle mitochondria of low-fat diet-fed mice receiving drug vehicle (LF-C), low-fat diet-fed mice receiving mitochondria-targeted vitamin E derivative (MitoVit E) (LF-E), medium-fat diet-fed mice receiving drug vehicle (MF-C) and medium-fat diet-fed mice receiving MitoVit E (MF-E)
(Mean values with their standard errors for seven to eight mice per group)
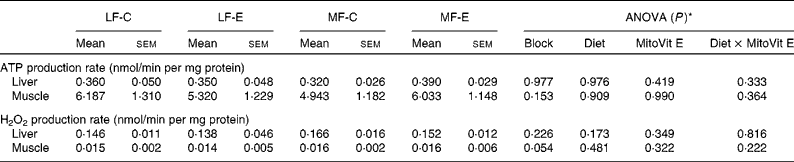
* Values of ANOVA (P) are P values for block effect and main effects, such as diet, MitoVit E and interaction between diet and MitoVit E in the LF-C, LF-E, MF-C and MF-E groups.
Measurement of mitochondrial number and size
The number and average size of the intermyofibrillar mitochondria were not affected by MitoVit E treatment in either LF or MF diet-fed mice (Table 2). However, the number and average size of the subsarcolemmal mitochondria were four- and twofold higher in MF-E mice than in MF-C mice, respectively (P < 0·05). Conversely, the number of intermyofibrillar and subsarcolemmal mitochondria was significantly decreased in the MF-C group compared with the LF-C group (Table 2). In addition, there was a diet × MitoVit E interaction effect for the number of subsarcolemmal mitochondria (P = 0·035). The representative transmission electron microscopic images of subsarcolemmal and intermyofibrillar mitochondria from the soleus muscle of LF-C, LF-E, MF-C and MF-E mice are shown in Fig. 2, where arrows indicate mitochondria. Remarkably, in the MF-C group, very few intermyofibrillar mitochondria were observed, and subsarcolemmal mitochondria had an increased number of disarrayed cristae and a reduced electron density of the matrix. By contrast, MitoVit E administration increased the mitochondrial number and electron density in MF diet-fed mice.
Table 2 Mitochondrial number and average size in the soleus muscle of low-fat diet-fed mice receiving drug vehicle (LF-C), low-fat diet-fed mice receiving mitochondria-targeted vitamin E derivative (MitoVit E) (LF-E), medium-fat diet-fed mice receiving drug vehicle (MF-C) and medium-fat diet-fed mice receiving MitoVit E (MF-E) mice
(Mean values with their standard errors, analysis of five images per mouse and two mice per group)
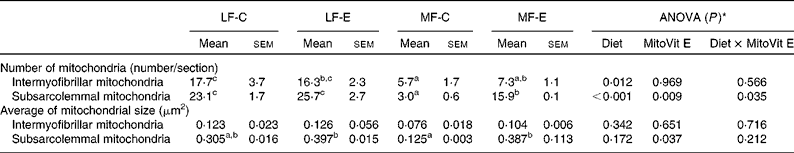
a,b,c Mean values within a row with unlike superscript letters were significantly different (P < 0·05).
* Values of ANOVA (P) are P values for main effects, such as diet, MitoVit E and interaction between diet and MitoVit E in the LF-C, LF-E, MF-C and MF-E groups. There is no block effect in the ANOVA model because all the mice (n 8) for microscopy are from the same block.

Fig. 2 Representative transmission electron microscopic images (original magnification, 4000 × ) of subsarcolemmal and intermyofibrillar mitochondria from the soleus muscle of low-fat diet-fed mice receiving drug vehicle (LF-C), low-fat diet-fed mice receiving mitochondria-targeted vitamin E derivative (MitoVit E) (LF-E), medium-fat diet-fed mice receiving drug vehicle (MF-C) and medium-fat diet-fed mice receiving MitoVit E (MF-E). Arrows indicate mitochondria.
Measurement of urinary isoprostane after treatments
After 2 and 4 weeks of HF feeding, urinary isoprostane concentration increased four- and fivefold, respectively (Fig. 1(a)). However, after the mice were switched from the HF diet to either the LF or the MF diet with or without MitoVit E, urinary isoprostane concentration decreased in the first week (week 6) immediately (Table 3). The levels of the decreased urinary isoprostane after the first week (week 6) were maintained for the rest of the study (weeks 7–10) for all four groups (Table 3). The administration of MitoVit E significantly enhanced the decrease in urinary isoprostane in LF diet-fed mice (LF-C v. LF-E) for the whole study, but not in MF diet-fed mice (MF-C v. MF-E) except at week 6 (Table 3). In addition, the decrease in urinary isoprostane was significantly higher in the LF-E group than in the other three groups during the whole study (weeks 6–10), indicating that the combination of LF diet and MitoVit E administration is most effective in decreasing systemic oxidative stress.
Table 3 Decrease in urinary isoprostane from low-fat diet-fed mice receiving drug vehicle (LF-C), low-fat diet-fed mice receiving mitochondria-targeted vitamin E derivative (MitoVit E) (LF-E), medium-fat diet-fed mice receiving drug vehicle (MF-C) and medium-fat diet-fed mice receiving MitoVit E (MF-E)*
(Mean values with their standard errors for six mice per group)
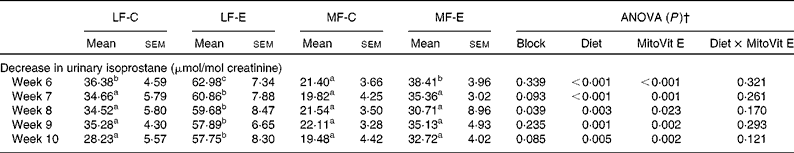
a,b,c Mean values within a row with unlike superscript letters were significantly different (P < 0·05).
* Urinary isoprostane was normalised by urinary creatinine, and decrease in urinary isoprostane was calculated by subtracting the levels of urinary isoprostane at weeks 6, 7, 8, 9 and 10 from the baseline level of urinary isoprostane at week 5, respectively.
† Values of ANOVA (P) are P values for block effect and main effects, such as diet, MitoVit E and interaction between diet and MitoVit E in the LF-C, LF-E, MF-C and MF-E groups.
Measurement of redox parameters in the plasma
Plasma SOD activity was lower in the MF-C group than in the LF-C group (P < 0·05). Plasma GPx activity was also significantly decreased in the MF-E group compared with the LF-E group (Table 4). Concentrations of plasma H2O2 in the LF-C and LF-E groups were significantly lower than in the MF-C and MF-E groups, respectively (Table 4). A significant increase in plasma SOD activity was observed in MF-E group when compared with the MF-C group. In addition, there was a diet × MitoVit E interaction effect for plasma SOD activity (P = 0·038).
Table 4 Plasma parameters of low-fat diet-fed mice receiving drug vehicle (LF-C), low-fat diet-fed mice receiving mitochondria-targeted vitamin E derivative (MitoVit E) (LF-E), medium-fat diet-fed mice receiving drug vehicle (MF-C) and medium-fat diet-fed mice receiving MitoVit E (MF-E)*
(Mean values with their standard errors for seven to ten mice per group)
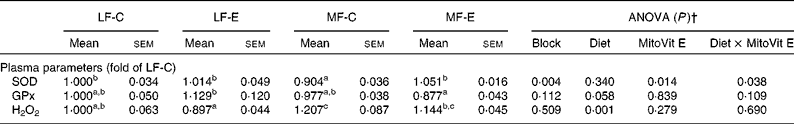
SOD, superoxide dismutase; GPx, glutathione peroxidase.
a,b,c Mean values within a row with unlike superscript letters were significantly different (P < 0·05).
* Mean values are fold changes relative to the LF-C group set to 1 unit.
† Values of ANOVA (P) are P values for block effect and main effects, such as diet, MitoVit E and interaction between diet and MitoVit E in the LF-C, LF-E, MF-C and MF-E groups.
Discussion
Our oxidative stress model was established by feeding mice a HF diet for 5 weeks to induce higher oxidative stress, which was indicated by the increase in urinary isoprostane (Fig. 1(a)). Cumulative evidence strongly suggests that elevated oxidative stress is associated with obesity development(Reference Keaney, Larson and Vasan36–Reference Ozata, Mergen and Oktenli39). This suggestion has been confirmed by our observations that urinary isoprostanes increased with increased body weight during the first 4 weeks of HF feeding (Fig. 1(a) and (b)).
Mitochondrial ATP production rate was not affected by MitoVit E in either the liver or the gastrocnemius. This observation was confirmed by using different combinations of substrates for measuring the mitochondrial ATP production rates, such as glutamate plus malate in the present study, glutamate plus malate plus palmitoyl-l-carnitine(Reference Mao, Kraus and Kim40) and glutamate plus malate plus palmitoyl-l-carnitine plus α-ketoglutarate (G Mao, GA Kraus, I Kim, ME Spurlock, TB Bailey and DC Beitz, unpublished results).
No effects of MitoVit E on mitochondrial H2O2 production rate were observed in the present study when succinate was used as the substrate. However, when glutamate plus malate was used as the substrate, MitoVit E in the same study significantly decreased the mitochondrial H2O2 production in the liver but not in the muscle of MF diet-fed mice(Reference Mao, Kraus and Kim40), indicating that the effect of MitoVit E on scavenging mitochondria-derived ROS might be substrate-dependent and thus complex-dependent.
In addition, the capacity of MitoVit E in reducing the mitochondrial oxidative damage has been confirmed by our observation that it significantly reduced protein carbonyl in the liver mitochondria of MF-E mice compared with MF-C mice(Reference Mao, Kraus and Kim40), which is consistent with the previous finding that MitoVit E protected liver mitochondria from Fe/ascorbate-induced oxidative damage by decreasing protein carbonyl(Reference Smith, Porteous and Coulter41).
A previous study has found that in a diabetic mouse model after 16 weeks of HF, high-sucrose feeding, the number and size of both subsarcolemmal and intermyofibrillar mitochondria in the skeletal muscle decreased along with a significant increase in ROS production(Reference Bonnard, Durand and Peyrol42). The same study also reported that mice treated with streptozotocin, a well-known model of hyperglycaemia-associated oxidative stress without insulin resistance and obesity, significantly decreased the mitochondrial number, whereas N-acetylcysteine treatment of these streptozotocin mice restored the mitochondrial density, indicating that mitochondrial number and size might be altered by oxidative stress. In addition, a decrease in mitochondrial size and number has also been reported in the neurons of patients with Alzheimer's disease(Reference Baloyannis43), the pathogenesis of which has been extensively attributed to mitochondrial oxidative damage and dysfunction(Reference Moreira, Carvalho and Zhu44).
Consistent with this concept, the present results showed that the MF diet decreased the number of intermyofibrillar and subsarcolemmal mitochondria in the soleus muscle, whereas MitoVit E increased the number and size of the subsarcolemmal mitochondria. However, why MitoVit E only has the effect on the subsarcolemmal but not on the intermyofibrillar mitochondria still remains elusive. Combined with H2O2 production rate data in the gastrocnemius though, another question arises: why did MitoVit E have no effect on mitochondrial H2O2 production rate in the gastrocnemius, but it could still increase the size and number of subsarcolemmal mitochondria in the soleus? We could not exclude the possibility that MitoVit E might affect the subsarcolemmal mitochondrial density by some other unknown properties other than as an antioxidant. Alternatively, this question could also have to do with the different fibre types in the two muscles (red oxidative soleus v. white glycolytic gastrocnemius). The precise mechanism still remains unclear until the effects of MitoVit E on the mitochondrial density in the gastrocnemius and the mitochondrial H2O2 production rate in the soleus muscle are investigated in the future.
Clinically, one of the keys to success in the diagnosis and management of those oxidative damage-induced metabolic disorders is the utilisation of those easily accessible plasma- and urine-based biomarkers for the objective assessment of oxidative stress and response to therapeutics.
In the present study, urinary isoprostane was used as a systematic oxidative stress marker. To our knowledge, the effect of MitoVit E on plasma or urinary isoprostanes has never been studied before. However, there are several studies available in the literature for the effect of non-targeted vitamin E on the isoprostanes, even though their results are not consistent. Both α-tocopherol and mixed tocopherol supplementation resulted in reduced plasma isoprostanes but did not affect 24 h urinary isoprostanes in patients with type 2 diabetes(Reference Wu, Ward and Indrawan45). However, in the patients with coronary disease, high-dose α-tocopherol supplementation significantly reduced urinary isoprostanes(Reference Devaraj, Tang and Adams-Huet46). By contrast, vitamin E supplementation failed to decrease urinary isoprostanes in either lean or obese mice(Reference Hasty, Gruen and Terry47).
The present results indicate that MitoVit E decreased urinary isoprostanes, and its effect was more significant in LF diet-fed mice than in MF diet-fed mice, which could be attributed to the fact that the decrease in urinary isoprostane concentration in the LF-E group was significantly higher than in the other three groups (LF-C, MF-C and MF-E).
Decrease in antioxidant status leading to the generation of oxidative stress may play an important role in the pathogenesis of oxidative stress-induced metabolic disorders(Reference Stanek, Cieslar and Romuk48). Indeed, decreased activities of plasma SOD and GPx were commonly observed in different models of oxidative stress, whereas antioxidant treatments normalised their activities(Reference Shen, Tang and Wu49–Reference Fernandez-Pachon, Berna and Otaolaurruchi51).
Consistently, the present results indicate that the MF diet significantly decreased plasma SOD and GPx activities and thus increased plasma H2O2 concentration when compared with the LF diet. However, in the present study, MitoVit E only increased the activity of plasma SOD but not the activity of plasma GPx. Similar observations were found in a lipopolysaccharide-induced oxidative stress model in the rat liver(Reference Sebai, Sani and Yacoubi50). Lipopolysaccharide treatment significantly decreased hepatic antioxidant enzyme activities such as SOD and GPx, whereas pre-treatment with resveratrol, a polyphenol with antioxidant properties abundantly found in red wine, significantly reversed lipopolysaccharide-induced decrease in SOD activity but only slightly (and not significantly) improved the lipopolysaccharide-induced decrease in GPx activity(Reference Sebai, Sani and Yacoubi50). By contrast, in the ethanol-induced oxidative stress model, MitoVit E has been reported to mitigate ethanol-induced suppression of GPx/glutathione reductase functions, protein expression of γ-glutamylcysteine synthetase and total cellular glutathione levels(Reference Siler-Marsiglio, Pan and Paiva23). Whether MitoVit E might also alter the levels of oxidised/reduced glutathione and γ-glutamylcysteine synthetase in our model is still unknown and needs to be studied in the future.
In summary, the present study found that MitoVit E did not affect mitochondrial ATP or H2O2 (when succinate was the substrate) production rates in both the liver and the gastrocnemius. However, MitoVit E increased the number and average size of the subsarcolemmal, though not the intermyofibrillar, mitochondria in the soleus muscle of MF diet-fed mice. In addition, the combination of the LF diet and MitoVit E administration decreased the urinary isoprostane concentration most effectively. Finally, MitoVit E increased plasma SOD but not GPx activity in MF diet-fed mice.
Acknowledgements
The authors acknowledge Dr Harry Horner and Randy Denadel for helping in the transmission electron microscopy measurements, Yuhong Liu for valuable suggestions in the mitochondrial preparation and measurements of the mitochondrial ATP and H2O2 production rates, and Dr Lorene Leiter and Peter Scarbrough for the critical reading and correction of the manuscript. The present study was supported by the Nutrition and Wellness Research Center/USDA at Iowa State University. G. M. conducted the study, collected and analysed the data, and drafted the manuscript. G. A. K. and I. K. synthesised MitoVit E. M. E. S. and D. C. B. designed and supervised the study. T. B. B. provided the statistical assistance. All authors read and approved the final manuscript. The authors have no conflicts of interest.







