


In pancreatic islet β-cells, ATP acts on ATP-dependent K+ channels leading to membrane depolarisation, opening of voltage-sensitive Ca2+ channels, increasing cytosolic Ca2+ concentration and stimulating insulin exocytosis(Reference Aguilar-Bryan, Nichols and Wechsler1). Under physiological conditions mitochondrial glucose metabolism provides most of the ATP required during insulin secretion(Reference Dyachok, Idevall-Hagren and Sågetorp2). Glucose metabolism leads to the enhancement of net tricarboxylic acid cycle (TAC) intermediates (TACI) including citrate, malate, oxaloacetate, α-ketoglutarate and succinate, a process known as anaplerosis(Reference MacDonald and Wheeler3, Reference MacDonald, Fahien and Brown4). The β-cell elevated TACI concentration has been associated with augmented oxidative energy production and insulin release(Reference Fransson, Rosengren and Schuit5, Reference MacDonald, Stoker and Hasan6). Despite the essential role of ATP, insulin release signalling by other metabolic coupling factors including malonyl-CoA, NADPH, leucine, glutamate and diacylglycerol has also been demonstrated, which suggests the existence of a complex process of insulin release regulation(Reference MacDonald, Fahien and Brown4, Reference MacDonald, Longacre and Stoker7). Metabolic coupling factors are generally derived from TACI extramitochondrial metabolism after their exit from the mitochondria, a process known as cataplerosis(Reference MacDonald, Fahien and Brown4). The TACI anaplerosis:cataplerosis ratio, therefore, plays a signalling role during insulin release in addition to contributing to enhanced ATP concentration.
Previous studies have demonstrated that pyruvate carboxylase (PC) activity and glucose-induced insulin secretion (GIIS) are strictly coupled, suggesting that an elevated anaplerosis–cataplerosis cycle is of substantial importance for insulin release(Reference Lu, Mulder and Zhao8). In 832/13 insulinoma cells treated with high concentrations of NEFA, GIIS is significantly reduced in association with a decreased mitochondrial anaplerosis–cataplerosis cycle(Reference Boucher, Lu and Burgess9).
Peripheral tissues with insulin resistance are expected to exhibit an impaired mitochondrial oxidative capacity followed by an elevated intracellular lipid content(Reference Kelley, Goodpaster and Wing10–Reference Phielix and Mensink13). Kelley et al. (Reference Kelley, Goodpaster and Wing10) examined the respiratory quotient in diabetic, obese and lean subjects. It was observed that in contrast to lean subjects, diabetic and obese patients were unable to switch from lipid to carbohydrate metabolism after 8 h following an insulin-stimulated glucose loading. Paradoxically, diabetic and obese individuals preferentially oxidised carbohydrate, demonstrating a poor capacity for lipid oxidation. These findings are in agreement with the proposition that insulin-resistant subjects exhibit impaired mitochondrial capacity and reduced fatty acid (FA) β-oxidation(Reference Petersen, Dufour and Befroy11, Reference Muoio and Newgard14). In addition, insulin-resistant subjects are expected to express elevated uncoupling protein-3 content, demonstrating a low mitochondrial capacity(Reference Schrauwen, Saris and Hesselink15, Reference Schrauwen16). In contrast, endurance-trained subjects exhibit elevated substrate consumption at rest and a high anaplerotic–cataplerotic capacity(Reference Befroy, Petersen and Dufour17). Chronic and acute exercise has also been shown to reduce insulin resistance(Reference Dubé, Amati and Stefanovic-Racic18–Reference Goodpaster, He and Watkins20). Under this condition, TACI anaplerosis is postulated to be required for adequate skeletal muscle contraction(Reference Bowtell, Marwood and Bruce21). Conversely, absence of an adequate TACI concentration leads to reduced muscle contraction tolerance, probably by a reduced mitochondrial oxidative flux(Reference Sahlin, Katz and Broberg22, Reference Thyfault23).
Intra-uterine and early postnatal protein malnutrition results in decreased insulin release(Reference Mann, Becker and Pimstone24–Reference Parra, Klish and Cuellar26) and pancreatic β-cell altered glucose metabolism(Reference Huang, Qiu and Shen27, Reference Benyshek, Johnston and Martin28). Our group has provided evidence that protein restriction reduces insulin secretion stimulated by glucose, amino acids, K+ and other secretagogues(Reference Latorraca, Reis and Carneiro29–Reference Filiputti, Ferreira and Souza32). This decreased capacity is related to alterations in gene expression, including mitogen-activated protein kinases, voltage-gated K+ channel and glucose transporters(Reference Araujo, Amaral and Filiputti31–Reference Delghingaro-Augusto, Ferreira and Bordin33). Adult offspring of pregnancy and early postnatal malnutrition develop peripheral insulin resistance(Reference Huang, Qiu and Shen27, Reference Benyshek, Johnston and Martin28, Reference Ozanne and Hales34) followed by reduced muscle contraction capacity(Reference Toscano, Manhães-de-Castro and Canon35), alterations that are closely related to type 2 diabetes development. Thus, there seems to be a narrow concert governed by mitochondrial function between β-cell insulin release and peripheral tissue metabolism.
Protein malnutrition has been reported to induce similar effects to obesity. Mitochondrial metabolism in pancreatic β-cells as well as peripheral tissues under this situation has been poorly investigated. We propose that protein malnutrition induces an imbalanced anaplerosis:cataplerosis ratio due to the reduced amino acid pool. So, the withdrawal of TACI would increase intracellular amino acid availability for protein synthesis, reducing TAC flux and consequently decreasing insulin release and sensitivity. The purpose of the present review is to report evidence of anaplerotic mechanisms involved with the regulation of insulin secretion, peripheral resistance and muscle contraction metabolism under ordinary situations and under protein malnutrition, suggesting future directions for research in this field.
Anaplerosis and insulin release
An increased blood glucose concentration stimulates pancreatic β-cells to secrete insulin but the molecular aspects concerning GIIS are not completely understood. Recent findings have proposed an oscillatory pattern of downstream events, leading to an increase of cytoplasmic Ca2+ levels, insulin granule docking and fusion with the plasma membrane, enhanced cyclic AMP production, amplified Ca signalling and activation of secretory mechanisms by Ca2+ ions(Reference Dyachok, Idevall-Hagren and Sågetorp2, Reference Jones, Fyles and Howell36–Reference MacDonald and Rorsman38). Several mechanisms have been proposed involving the KATP- and Ca2+-dependent pathway, the K+ATP-independent and Ca2+-dependent pathway, and possibly a K+ATP- and Ca2+-independent pathway(Reference Prentki39–Reference Gembal, Gilon and Henquin43). However, little is known about the metabolic regulation associated with these pathways.
Improved TACI anaplerosis–cataplerosis flux is needed to enhance intracellular ATP content(Reference MacDonald, Fahien and Brown4, Reference Fransson, Rosengren and Schuit5, Reference Ronnebaum, Ilkayeva and Burgess44). Although the mechanism involved remains unknown, a strong correlation has been documented between PC activity and GIIS(Reference Ronnebaum, Ilkayeva and Burgess44, Reference Curi, Carpinelli and Malaisse45). This enzyme, located in the mitochondrial matrix, catalyses the ATP-dependent carboxylation of pyruvate to form oxaloacetate(Reference Khan, Ling and Landau46). PC is highly expressed in the liver and kidney, and participates with phosphoenolpyruvate carboxykinase, fructose-1,6-bisphosphatase and glucose-6-phosphatase of gluconeogenesis(Reference Ronnebaum, Ilkayeva and Burgess44, Reference Curi47, Reference Schuit, De Vos and Farfari48). The lack of phosphoenolpyruvate carboxykinase activity and relatively low lipogenic capacity suggest an important role of PC in β-cells during oxidative energy metabolism. A proportion of about 40–50 % pyruvate enters β-cells during mitochondrial metabolism through PC reactions at stimulating glucose concentrations, a very high flux for a non-gluconeogenic tissue. The anaplerosis of glucose carbons, therefore, highly correlates with GIIS by β-cells(Reference MacDonald, Fahien and Brown4, Reference Schuit, De Vos and Farfari48). In INS cells (from the insulinoma cell line), anaplerosis was reported to be increased followed by a high basal insulin secretion after exposure to glucose. However, at a low glucose concentration, PC expression was demonstrated to be markedly decreased(Reference Lu, Mulder and Zhao8, Reference Jensen, Joseph and Ilkayeva49). Farfari et al. (Reference Farfari, Schulz and Corkey50) demonstrated that phenylacetic acid, a PC inhibitor, reduced GIIS in INS cells and pancreatic islets, an effect that was associated with reduced citrate accumulation. Similarly, Fransson et al. (Reference Fransson, Rosengren and Schuit5), examining the phenylacetic acid-induced effect on PC during insulin release in rat islets, demonstrated that anaplerosis via PC is required for an appropriated rise in the ATP:ADP ratio and insulin secretion. However, caution must be taken from the above studies once phenylacetic acid specificity is limited toward PC(Reference Ronnebaum, Ilkayeva and Burgess44). Very recently, Hasan et al. (Reference Hasan, Longacre and Stoker51), testing the hypothesis that anaplerosis via PC is important for GIIS in cell lines, verified that reduced expression of this enzyme using transfection of short hairpin RNA was associated with reduced PC activity and insulin release in response to glucose and other secretagogues. Although the mechanism remains to be elucidated, the influx of carbon intermediates into the TAC is critical for appropriate ATP generation in β-cells(Reference Curi, Carpinelli and Malaisse45). The proposed mechanisms have been extensively studied and supported by different metabolic pathways in which pyruvate is metabolised and/or recycled(Reference Ronnebaum, Ilkayeva and Burgess44, Reference Jensen, Joseph and Ilkayeva49, Reference Farfari, Schulz and Corkey50, Reference MacDonald52, Reference Guay, Madiraju and Aumais53). As in most mammalian tissues, pyruvate may follow two different routes, feeding the TAC with acetyl-CoA via pyruvate dehydrogenase and oxaloacetate via PC in pancreatic β-cells(Reference Curi, Carpinelli and Malaisse45).
Pyruvate dehydrogenase contributes with the TAC by the production of acetyl-CoA. In this reaction, one carbon from pyruvate is lost as CO2 and two are converted in the acetyl-CoA molecule, which will condensate with oxaloacetate to form citrate by citrate synthase (CS)(Reference MacDonald52). The oxaloacetate molecule is regenerated with the TACI, remaining constant at the expense of acetyl-CoA. Although much attention has been given to PC, the flux of pyruvate decarboxylation through pyruvate dehydrogenase in β-cells is estimated to be similar to that observed during pyruvate carboxylation by PC(Reference Guay, Madiraju and Aumais53). Sustained glucose supply raises the intracellular citrate content in β-cells, suggesting an association between PC and pyruvate dehydrogenase during the anaplerosis process(Reference Schuit, De Vos and Farfari48). However, the high K m of the CS for acetyl-CoA compared with oxaloacetate suggests that the flux of oxaloacetate from pyruvate via PC is greatly favoured, increasing the content of intermediates in the second span of TAC(Reference Srere54). Moreover, acetyl-CoA is a well known allosteric activator of PC, favouring the anaplerosis process and consequently the rate of intracellular ATP production(Reference Newsholme and Leech55). However, the elevated TACI content exerts an inhibitory effect on most of the regulatory sites from the TAC(Reference MacDonald, Fahien and Brown4, Reference Newsholme and Leech55, Reference MacDonald, Fahien and Buss56), which will further allow the TACI export from the mitochondria to the cytosol. Once accumulated, TACI not only provide ATP through the mitochondrial electron transport chain but also are exported (cataplerosis) to the cytosol(Reference MacDonald, Fahien and Brown4). In this latter process, carbon derived from pyruvate carboxylation exits from the mitochondria to the cytosol, primarily as malate(Reference MacDonald, Fahien and Brown4, Reference Guay, Madiraju and Aumais53). In the cytosol, malate is converted to pyruvate in a NADPH generation reaction catalysed by cytosolic malic enzyme (MEc), which can be transported back to the mitochondria(Reference MacDonald, Fahien and Brown4). This cycle, also known as the pyruvate–malate shuttle, occurs inside the mitochondrial matrix and exerts a critical role in β-cells during both the anaplerosis and cataplerosis processes by recycling pyruvate and NADPH production, a potent metabolic coupling factor(Reference MacDonald52, Reference Guay, Madiraju and Aumais53) (Fig. 1). Evidence of pyruvate–malate shuttle activity was recently demonstrated during GIIS in INS cells. The inhibition of the malate dicarboxylate transporter by pharmacological inhibition and/or by RNA interference (RNAi) markedly reduced the GIIS(Reference Guay, Madiraju and Aumais53).
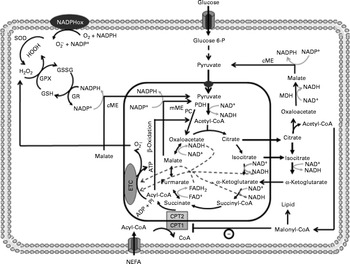
Fig. 1 Anaplerosis and cataplerosis pathways during insulin secretion in β-cells. The pyruvate cycling is the main cytosolic site of NADPH production. NADPH is one of the most import antioxidants in β-cells. This anaplerotic process includes the pyruvate–malate shuttle, pyruvate–citrate cycle and pyruvate–isocitrate–α-ketoglutarate cycle. NADPHox, NADPH–oxidase enzymic complex; SOD, superoxide dismutase; O2− ∙, superoxide anion; GSSG, oxidised glutathione; GPX, glutathione peroxidase; GSH, glutathione; GR, glutathione reductase; cME, cytosolic malic enzyme; glucose 6-P, glucose 6-phosphate; MDH, malate dehydrogenase; CPT, carnitine palmitoyltransferase; mME, mitochondrial malic enzyme; PDH, pyruvate dehydrogenase; PC, pyruvate carboxylase; ETC, electron transport chain; Pi, inorganic phosphate.
Although the export of citrate is not increased as much as malate, during β-cell anaplerotic activity, citrate content has also been shown to suffer a larger range of oscillation compared with other TACI, an effect that was correlated with ATP and NAD(P)H oscillations(Reference MacDonald, Fahien and Buss56). Elevated malate and citrate concentrations suggest that the K m of malate dehydrogenase and CS for oxaloacetate must be of similar magnitude, favouring rapid TAC expansion(Reference Ronnebaum, Ilkayeva and Burgess44, Reference MacDonald52, Reference Guay, Madiraju and Aumais53). Similarly to malate, citrate after exiting the mitochondria is converted by ATP–citrate lyase to oxaloacetate and acetyl-CoA(Reference Ronnebaum, Ilkayeva and Burgess44, Reference Guay, Madiraju and Aumais53). Oxaloacetate in a sequence of two reactions is reduced to malate by malate dehydrogenase, which will further be converted to pyruvate by MEc. In this reaction, NADP+ is used as cofactor favouring the rise of cytosolic NADPH:NADP+ and pyruvate transport back to the mitochondria. This cycle is defined as the pyruvate–citrate cycle(Reference Ronnebaum, Ilkayeva and Burgess44, Reference Farfari, Schulz and Corkey50, Reference Guay, Madiraju and Aumais53) (Fig. 1). Recent studies have demonstrated that oscillatory ATP production is strongly associated with cytosolic citrate content(Reference MacDonald, Fahien and Buss56). This relationship suggests that citrate might be by itself coordinating the activities of TAC and anaerobic glycolysis. Inside the mitochondrial matrix, citrate has been described as an important inhibitor of CS(Reference Newsholme and Leech55). This effect will favour its mitochondrial accumulation and consequently its exit toward the cytosol. Once accumulated into the cytosol, citrate might inhibit glycolysis via allosteric modulation of phosphofructokinase(Reference Newsholme and Leech55, Reference Tornheim, Andrés and Schultz57). This mechanism, associated with elevated cytosolic NADPH:NADP+, could explain, at least in part, the oscillatory pattern of insulin secretion in β-cells.
An increased NADPH:NADP+ inhibits the pentose phosphate pathway. Although this route is described as exhibiting low activity in β-cells, inhibition of the pentose phosphate pathway might exert a metabolic-saving effect, driving the glucose-6-phosphate to glycolysis(Reference Farfari, Schulz and Corkey50). On the other hand, acetyl-CoA is converted to malonyl-CoA by acetyl-CoA carboxylase, a well-known inhibitor of FA metabolism(Reference Coyle, Hargreaves and Thompson58, Reference Silveira, Fiamoncini and Hirabara59). In a tissue with elevated anaplerosis capacity, the cytosolic NADH:NAD+ ratio is reduced, allowing sustained glyceraldehyde 3-phosphate dehydrogenase activity and glycolytic flux, leading to an increase in the ATP:ADP ratio(Reference MacDonald, Fahien and Brown4, Reference Newsholme and Leech55). In β-cells, glucose oxidation provides a higher mitochondrial electrochemical gradient (Δψ) and ATP:ADP ratio than FA(Reference Willis and Jackman60), whereas FA oxidation seems to induce mitochondrial uncoupling and reduced ATP synthesis(Reference Spacek, Santorová and Zacharovová61). There is strong evidence linking PC activity and the pyruvate–citrate cycle with insulin secretion(Reference Fransson, Rosengren and Schuit5, Reference Lu, Mulder and Zhao8, Reference Guay, Madiraju and Aumais53, Reference Cline, Lepine and Papas62). PC inhibition resulted in decreased GIIS, which was positively correlated with citrate concentration(Reference Farfari, Schulz and Corkey50, Reference Hasan, Longacre and Stoker51). Furthermore, impairment of mitochondria citrate metabolism by either inhibition of mitochondrial di- and tricarboxylate carriers or deletion of citrate lyase and MEc genes leads to reduced GIIS, with no change in glucose oxidation, probably by a reduction in malonyl-CoA and NADPH synthesis(Reference Guay, Madiraju and Aumais53).
The importance of the anaplerotic PC–pyruvate–citrate cycle pathway was reinforced when β-cells were incubated in the presence of weak insulin secretagogues including acetoacetate, β-hydroxybutyrate, monometyl succinate and lactate. When incubated together, these metabolites increased acetyl-CoA, oxaloacetate and consequently citrate concentrations, that were able to enhance insulin release by 10- to 20-fold, almost the same effect observed for GIIS(Reference MacDonald, Longacre and Stoker7). However, a strong correlation between GIIS and a non-PC-derived anaplerosis pathway in insulinoma cell lines was observed, suggesting that other pathways, probably aspartate aminotransferase, act in combination with PC in TAC anaplerosis-induced GIIS, enhancing oxaloacetate levels through aspartate and α-ketoglutarate consumption, respectively(Reference Simpson, Khokhlova and Oca-Cossio63). MacDonald et al. (Reference MacDonald, Stoker and Hasan6) demonstrated that incorporation of carbon from pyruvate into lipids was not lowered in the citrate lyase-deficient INS-1 cell line, suggesting that citrate is not the only cataplerotic carbon carrier from the mitochondria to cytosol. Further studies are, therefore, needed to clarify other anaplerotic and cataplerotic pathways and substrates that regulate GIIS.
Alternatively, pyruvate cycling during TACI expansion can also occur via pyruvate, isocitrate and α-ketoglutarate(Reference Ronnebaum, Ilkayeva and Burgess44). Unlike the pyruvate–malate shuttle, oxaloacetate formed by PC is converted to citrate and isocitrate, which then leave the mitochondria forming oxaloacetate and acetyl-CoA through citrate lyase or α-ketoglutarate through cytosolic NADP+-dependent isocitrate dehydrogenase (ICDc). The pyruvate cycling can take place via conversion of oxaloacetate to malate and further to pyruvate by malate dehydrogenase and MEc, respectively(Reference Ronnebaum, Ilkayeva and Burgess44, Reference Jensen, Joseph and Ilkayeva49, Reference Guay, Madiraju and Aumais53). In addition, α-ketoglutarate can re-enter the mitochondria forming malate by following the TAC reactions, which will be converted to pyvurate via MEc or mitochondrial malic enzyme with elevated NADPH production (Fig. 1). Ronnebaum et al. (Reference Ronnebaum, Ilkayeva and Burgess44), examining the role of ICDc in control of GIIS in β-cells using ICDc iRNA, observed that suppression of ICDc attenuated the glucose-induced increment in pyruvate cycling and intracellular NADPH content. These findings, therefore, suggest that the pyruvate cycling pathway involving ICDc plays an important role in the control of GIIS.
In addition to the pyruvate cycling shuttle enzymes, the role of glutamate dehydrogenase (GDH) during anaplerosis-induced insulin release has recently been reviewed(Reference Maechler, Carobbio and Rubi64). Mice with overexpression of GDH had enhanced insulin release(Reference Carobbio, Ishihara and Fernandez-Pascual65). Carobbio et al. (Reference Carobbio, Frigerio and Rubi66), using GDH knockout rats, showed a near 37 % reduction in GIIS. Studies using leucine, a GDH-positive allosteric modulator, as an insulin secretagogue, showed enhanced insulin release(Reference Filiputti, Ferreira and Souza32, Reference Zawalich, Yamazaki and Zawalich67). To date, it is not known if GDH acts as an anaplerotic enzyme producing α-ketoglutarate or has a cataplerotic function generating glutamate at the expense of α-ketoglutarate during insulin release. Accumulated evidence has shown the link between GDH and GIIS(Reference Maechler, Carobbio and Rubi64). In addition to the possible anaplerotic and cataplerotic role during insulin release, GDH produces NADPH, another important metabolic coupling factor(Reference MacDonald, Fahien and Brown4).
Anaplerosis, intracellular redox status and insulin release in β-cells
Interestingly, most alterations in anaplerotic- and cataplerotic-related mechanisms are involved with NADPH production (Fig. 2). As first reported by MacDonald(Reference MacDonald52), the pyruvate cycling may be the main cytosolic site of NADPH production with much higher capacity than the pentose phosphate pathway in β-cells. In this sense, the cataplerosis of malate and citrate would increase NADPH production. The β-cells’ needs for this reducing agent are still not known. However, NADPH is involved in FA synthesis and cellular redox modulation. As β-cells exhibit a relatively low rate of FA synthesis(Reference Brun, Roche and Assimacopoulos-Jeannet68), the main NADPH function could be attributed to the regulation of intracellular redox status. Also, one would speculate that the low pentose phosphate pathway activity must be of relevance for β-cells, as this pathway is highly associated with lipid synthesis in different tissues. However, a high intracellular lipid content might be potentially toxic for β-cells, in which an elevated anaplerosis and cataplerosis process is required during GIIS.
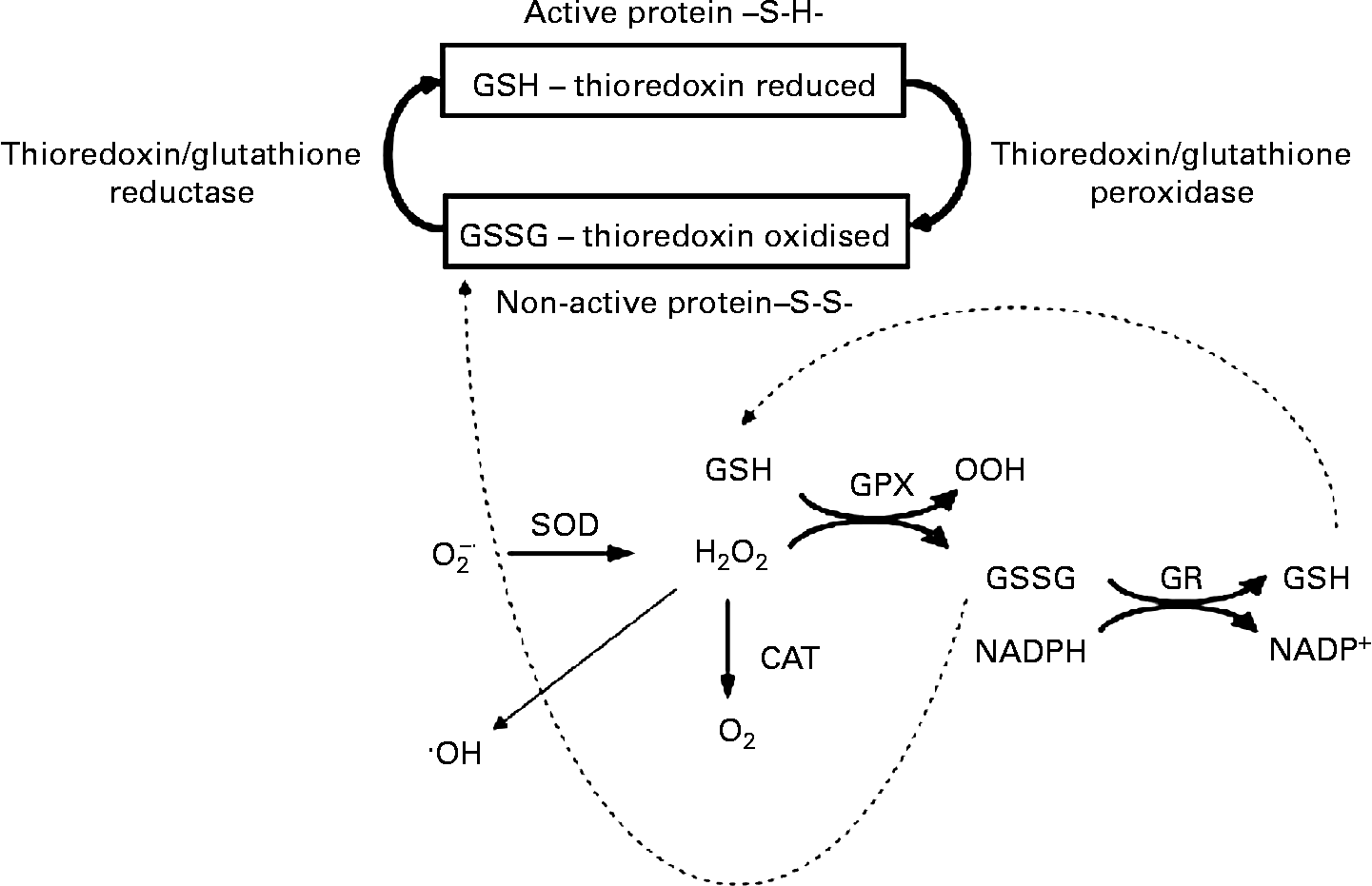
Fig. 2 The β-cell antioxidant system. The NADPH production during anaplerosis provides a substrate for glutathione and thioredoxin systems, which in turn will favour the intracellular redox status. GSH, glutathione; GSSG, oxidised glutathione; SOD, superoxide dismutase; O2− ∙, superoxide anion; ∙OH, hydroxyl radical; GPX, glutathione peroxidase; GR, glutathione reductase; CAT, catalase; S-S, thiol and disulfide (-S-S-).
Redox alteration of β-cells to a more reduced status is needed for proper insulin secretion(Reference Ramirez, Rasschaert and Sener69). The NADP+:NADPH ratio seems to play an important role in the redox modulation of insulin release, since NADPH is the substrate for several pro- and antioxidant enzymes(Reference MacDonald, Fahien and Brown4). The expansion of TACI during GIIS, therefore, seems to be important not only for energy production but also to regulate the intracellular redox balance.
During basal secretion, β-cells present a relatively low antioxidant capacity(Reference Newsholme, Haber and Hirabara70), but acute glucose loading leads to a fast induction of superoxide dismutase and glutathione peroxidase (GPX) activities, indicating an elevated production of reactive oxygen species (ROS)(Reference Oliveira, Curi and Carpinelli71, Reference Poitout and Robertson72). Glutathione reductase and thioredoxin, two NADPH-consuming enzymes, together with glutaredoxin are highly expressed in pancreatic islets(Reference Nagaoka, Iuchi and Ikeda73, Reference Ivarsson, Quintens and Dejonghe74), indicating the importance of reduced glutathione turnover in these cells. These findings, therefore, suggest that NADPH production during the export of TACI to the cytosol modulates Ca2+-dependent insulin secretion by regulating the intracellular redox balance(Reference Ivarsson, Quintens and Dejonghe74, Reference Reinbothe, Ivarsson and Li75). Oliveira et al. (Reference Oliveira, Verlengia and Carvalho76) also showed that β-cells express phagocyte-like NAD(P)H oxidase, a NADPH-consuming enzyme, which is known to produce ROS. Although the physiological role of β-cell NAD(P)H oxidase remains unclear, its activity may play a role, as demonstrated in other cell types, during Ca2+ release and consequently insulin secretion(Reference Hu, Zheng and Zweier77). Indeed, Morgan et al. (Reference Morgan, Rebelato and Abdulkader78) provided primary evidence for β-cell NAD(P)H oxidase-induced ROS production regulation of glucose flux and oxidation as well as Ca2+ intracellular response.
Robertson & Harmon(Reference Robertson and Harmon79) described the importance of GPX for the control of β-cell redox status. This enzyme acts in combination with glutathione reductase. Under low NADPH content, GPX has its ROS detoxification capacity markedly compromised. NO has also been proposed to have a protective effect upon the endoplasmic reticulum under ROS-induced stress(Reference Kitiphongspattana, Khan and Ishii-Schrade80). In addition, NO has been reported to stimulate insulin gene transcription(Reference Campbell, Richardson and Ferris81). NO is synthesised by NO synthase, which also uses NADPH as a substrate. The redox imbalance leading to β-cell oxidative stress has been implicated in several dysfunctions including low insulin release, cell proliferation and death, which are directly related to type 2 diabetes development(Reference Newsholme, Haber and Hirabara70).
Protein malnutrition, β-cell molecular alterations and insulin release
Protein-deficient diets lead to impaired insulin secretion in response to oral or intravenous glucose infusion and to other secretagogues(Reference Mann, Becker and Pimstone24–Reference Parra, Klish and Cuellar26, Reference Latorraca, Reis and Carneiro29–Reference Filiputti, Ferreira and Souza32, Reference Weinkove, Weinkove and Pimstone82–Reference Leon-Quinto, Magnan and Portha85). Under such conditions, the expression of pancreatic and duodenal homeobox-1, a transcription factor that plays a role in the maintenance of β-cell homeostasis, is markedly reduced in association with reduced pancreatic islet area and insulin release(Reference Arantes, Teixeira and Reis86). Expression of signalling proteins, such as protein kinase A and protein kinase C, is also reduced during protein malnutrition(Reference Ferreira, Filiputti and Arantes87, Reference Ferreira, Barbosa and Stoppiglia88). Several genes involved in insulin production and secretion mechanisms also have their expression altered(Reference Delghingaro-Augusto, Ferreira and Bordin33). Recently, it was demonstrated that Ca2+ uptake and insulin mRNA content were also reduced in undernourished rats, leading to reduced insulin release in response to glucose(Reference Latorraca, Carneiro and Mello89, Reference de Barros Reis, Arantes and Cunha90). These outcomes might in part be attributed to the reduced expression of both constitutive and inducible NO synthase isozymes under protein malnutrition(Reference Wu, Flynn and Flynn91). NO has been demonstrated to protect against endoplasmic reticulum stress and has a stimulating effect upon insulin gene transcription under regular fed state(Reference Kitiphongspattana, Khan and Ishii-Schrade80, Reference Campbell, Richardson and Ferris81). In addition, β-cells from rats fed with a protein-deficient diet have decreased expression of protein kinase B (PKB or Akt), mammalian target of rapamycin (mTOR) and p70s6k(Reference Filiputti, Ferreira and Souza32).
Although substrate availability might be reduced, capacity for oxidative ATP synthesis seems to be enhanced in protein deficiency. This statement is based on the findings of an almost threefold increase in ATP-synthase F1 complex expression, whereas GLUT-2 expression in β-cells was decreased(Reference Delghingaro-Augusto, Ferreira and Bordin33). The rise in ATP synthase gene expression may reflect an adaptation to low substrate availability, reduced mitochondrial metabolism and anaplerotic capacity. In contrast, the content of β-cell GLUT-1 and GLUT-2, intracellular glucose availability and glycolytic flux in fetuses from undernourished rats and adult undernourished rats were not different from control. However, mitochondrial glucose oxidation was found to be directly related to pancreatic and duodenal homeobox-1 expression and insulin secretion in undernourished rats, providing a possible link between metabolic and molecular mechanisms of insulin production and secretion in protein malnutrition(Reference Martín, Fernández and Pascual-Leone92).
Protein malnutrition, anaplerosis and insulin release
Concerning metabolic aspects of protein malnutrition-induced reduction in insulin release, Sener et al. (Reference Sener, Reusens and Remacle93) have shown that low-protein-fed rats have reduced glucose oxidation as demonstrated by a decreased metabolite flux through the glycerol phosphate shuttle in β-cells from malnourished rats, probably by a low mitochondrial FAD-linked glycerophosphate dehydrogenase activity(Reference Rasschaert, Reusens and Dahri94). In addition, at high glucose concentration, low-protein-fed rats showed an elevated glycolytic flux. However, leucine transamination to α-ketoisocaproic acid and further production of α-ketoglutarate was significantly reduced, indicating a poor anaplerotic capacity(Reference Sener, Reusens and Remacle93).
The impaired insulin release in malnourished rats might be, therefore, related to the lower mitochondrial oxidative and anaplerotic capacity (Fig. 3). The reduced β-cell anaplerosis in malnutrition is supported by findings that insulin synthesis is also regulated by succinate and/or succinyl-CoA cataplerosis(Reference Alarcon, Wicksteed and Prentki95, Reference Attali, Parnes and Ariav96). This finding is in accordance with reduced β-cell insulin mRNA levels in undernourished rats(Reference de Barros Reis, Arantes and Cunha90, Reference Martín, Fernández and Pascual-Leone92). Moreover, malonyl-CoA content, another insulin release signalling molecule, has been shown to be reduced under protein malnutrition(Reference de Barros Reis, Arantes and Cunha90). Malonyl-CoA is produced through mitochondrial citrate cataplerosis, and one of its actions is to limit FA oxidation, stimulating GIIS(Reference MacDonald, Fahien and Brown4). There seems to be a pyruvate–citrate cycle impairment under protein malnutrition, since intracellular malonyl-CoA content is very low. Thus, PC expression might be altered under this situation and the reasons are still unknown. Aspartate aminotransferase is identified as another main anaplerotic pathway under physiological conditions(Reference Simpson, Khokhlova and Oca-Cossio63). However, it has not been investigated in protein restriction conditions. Under physiological conditions, glutamate is described as a non-essential anaplerotic substrate(Reference Simpson, Khokhlova and Oca-Cossio63) but, under protein malnutrition, this amino acid may exert a role during anaplerosis. The hypothesis for a main glutamate anaplerotic route is based on the higher activity of glutamate–pyruvate transaminase shown in undernourished rats(Reference Rasschaert, Reusens and Dahri94). In this condition, an increased insulin release is observed in response to leucine stimulation, a positive GDH allosteric modulator(Reference Filiputti, Ferreira and Souza32). One possible reason could be an alternative pathway for pyruvate, α-ketoglutarate and NADPH synthesis. In agreement, we have recently demonstrated that GDH protein expression is reduced in rats submitted to protein undernourishment. However, leucine supplementation restored GDH expression as well as GIIS to control levels(Reference Silva, Filiputti and Zoppi97), probably by enhanced α-ketoglutarate and NADPH production.
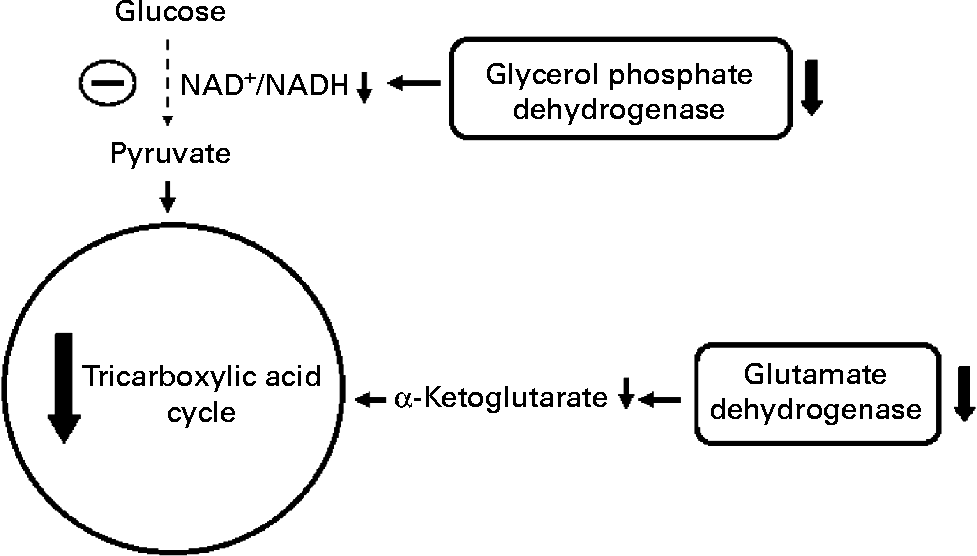
Fig. 3 Effect of protein malnutrition on anaplerosis and cataplerosis pathways during insulin secretion in β-cells. The pyruvate cycling including the pyruvate–malate shuttle, pyruvate–citrate cycle and pyruvate–isocitrate–α-ketoglutarate cycle is severely affected. Glucose anaplerosis might be affected by the decrease observed in FAD-linked glycerophosphate dehydrogenase activity. This observed decrease might impair mitochondrial reoxidation of cytosolic NADH, reducing glycolytic flux and pyruvate availability. The amino acid anaplerotic route seems to be also decreased by the reduced glutamate dehydrogenase expression which decreases α-ketoglutarate enhancement. Reduced anaplerotic capacity will result in lowered ATP:ADP and cataplerosis as well. Decreased cataplerosis flux would result in reduced metabolic coupling factors, such as malonyl-CoA and NADPH production. These metabolic alterations might impair insulin release.
Reusens et al. (Reference Reusens, Sparre and Kalbe98), examining the effect of a low-protein diet in pregnant rats, demonstrated that the gene expression of most TAC proteins was substantially up-regulated, an effect that was accompanied by a reduced expression of superoxide dismutase and heat shock protein-1, -1a and -1b. However, cytochrome c oxidase activity and ATP production were markedly reduced. Preliminary data from our laboratory provided evidence for a reduction of catalase activity, but GPX activity remained unchanged (APG Cappelli, CC Zoppi, A Trevisan, TM Batista, PMR da Silva and EM Carneiro, unpublished results). It seems reasonable, therefore, that during protein deficiency anaplerosis might be compromised, leading to reduced NADH:NAD+ and NADPH:NADP+ ratios and insulin synthesis and release. Further studies are needed to investigate the expression and content of anaplerotic enzymes as well as their substrate oscillations during insulin release events in undernourished β-cells.
Anaplerosis and peripheral insulin resistance
Skeletal muscle operates in a coordinated way with pancreatic islets in the control of glucose levels(Reference Phielix and Mensink13). The binding of insulin to its receptor induces insulin receptor tyrosine kinase activity. Tyrosine phosphorylation of insulin receptor substrate (IRS)-1 results in activation of the p85 regulatory subunit of phosphatidylinositol 3,4,5-trisphosphate (PI3) kinase and activates the p110 catalytic subunit, which increases phosphoinositides such as PI3. This leads to activation of phosphoinositide-dependent protein kinase and downstream PKB (Akt) and/or atypical protein kinase C(Reference Ishiki and Klip99). Phosphorylation of Akt substrate 160 (AS160), which has a GTPase-activating domain (Rab4), facilitates translocation of GLUT-4 to the sarcolemma, favouring glucose uptake(Reference Wei, Chen and Whaley-Connell100).
Intramuscular TAG (IMTG) accumulation leads to an increased concentration of FA metabolites including diacylglycerol, fatty acyl-CoA and ceramides that in turn activate serine kinase leading to IRS-1 phosphorylation of serine residues, and consequently inhibition of insulin downstream events(Reference Gual, Le Marchand-Brustel and Tanti101). In addition, higher levels of IMTG may increase ROS production and induce inflammation(Reference Wei, Chen and Whaley-Connell100, Reference Schenk, Saberi and Olefsky102).
Abnormal mitochondrial metabolism has been described in the insulin-resistant state(Reference Turner and Heilbronn12, Reference De Filippis, Alvarez and Berria103–Reference Rabøl, Boushel and Dela105). However, IMTG accumulation with no mitochondrial dysfunction has also been associated with the development of peripheral insulin resistance(Reference De Feyter, Van den Broek and Praet106–Reference Kraegen, Cooney and Turner109). Therefore, whether mitochondrial dysfunction is a cause or consequence of peripheral insulin resistance development is still unknown(Reference Phielix and Mensink13).
Interestingly, when sedentary obese insulin-resistant subjects were submitted to a moderate exercise training programme, insulin sensitivity was increased despite enhanced IMTG stores. However, this effect was associated with a reduction in diacylglycerol and ceramide content followed by an increased mitochondrial oxidative capacity(Reference Dubé, Amati and Stefanovic-Racic18). Similar results have been obtained in obese Zucker rats submitted to acute exercise. Despite an unchanged or increased concentration of intramuscular diacylglycerol and long-chain acyl-CoA, acute exercise improved insulin sensitivity, probably by the enhanced phosphorylation of AS160(Reference Thyfault23). Skeletal muscle contraction is a well-known stimulus to increase GLUT-4 translocation by an insulin-independent mechanism(Reference Thorell, Hirshman and Nygren110). Although the full mechanisms involved in contraction-induced GLUT-4 translocation are still unclear, sarcoplasmic Ca2+ efflux and the AS160 phosphorylation by the Akt–AMP-activated kinase pathway seem to play a pivotal role also in glucose uptake(Reference Cartee and Wojtaszewski111).
If mitochondrial dysfunction is not the main cause of insulin resistance, it can, at least in part, contribute to the development of this condition. A conciliatory hypothesis has been proposed that the link between mitochondrial metabolism and IMTG lipotoxicity-induced insulin resistance would be confined to the level of IMTG turnover inside muscle fibres. Thus, a mismatch between IMTG hydrolysis (lipolysis) and mitochondrial β-oxidation increases the intracellular lipid content with detrimental effects on insulin signalling and glucose metabolism. So, regular physical exercise plays a key role to protect against IMTG accumulation-induced insulin resistance(Reference Moro, Bajpeyi and Smith112).
The endproduct of FA β-oxidation is acetyl-CoA, and the further oxidation of this compound occurs inside the TAC. However, the first TAC reaction catalysed by CS is the condensation of acetyl-CoA with oxaloacetate, giving rise to citrate. Thus for a complete oxidation of FA molecules, a regular production of oxaloacetate is needed(Reference Siu, Donley and Bryner113). Muoio & Koves(Reference Muoio and Koves114) reviewed the molecular mechanism-induced mitochondrial metabolism dysfunction and proposed several mechanisms that may be involved in peripheral insulin resistance, including a reduced TAC flux. Indeed, type 2 diabetic patients have reduced TAC flux and consequently impaired complete FA oxidation(Reference Schrauwen and Hesselink115). In addition, reduced ADP phosphorylation in mitochondria from type 2 diabetic patients might result from reduced TAC and electron transport chain flux(Reference Phielix, Schrauwen-Hinderling and Mensink116).
One possible mechanism to explain the reduced TAC flux is an impaired anaplerotic capacity (Fig. 4). However, there are scarce data regarding anaplerotic pathways in insulin-resistant tissues. Befroy et al. (Reference Befroy, Petersen and Dufour17) examined whether resting skeletal muscle metabolism is altered in endurance-trained compared with sedentary subjects. These authors reported that trained subjects exhibit elevated substrate consumption at rest, suggesting a high anaplerotic and cataplerotic capacity. In contrast, elevated uncoupling protein-3 content has been demonstrated in insulin-resistant subjects, indicating an elevated IMTG content and consequently poor anaplerosis and cataplerosis capacity(Reference Schrauwen, Saris and Hesselink15, Reference Schrauwen16, Reference Schrauwen and Hesselink115).
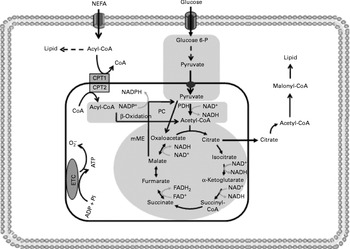
Fig. 4 Insulin resistance reduces tricarboxylic acid cycle (TAC) flux in peripheral tissues. The shadowed areas indicate the major pathways associated with low anaplerotic and cataplerotic capacity including glycolysis, the TAC and β-oxidation. Glucose 6-P, glucose 6-phosphate; CPT, carnitine palmitoyltransferase; PC, pyruvate carboxylase; PDH, pyruvate dehydrogenase; mME, mitochondrial malic enzyme; ETC, electron transport chain; O2− ∙, superoxide anion.
Reduced TAC flux can also be impaired by oxidative stress as previously reported in peripheral insulin resistance, a metabolic state which stimulates ROS production(Reference Silveira, Fiamoncini and Hirabara59, Reference Nicolson117–Reference Bonnard, Durand and Peyrol119). In fact, elevated concentrations of the superoxide anion might inhibit aconitase activity(Reference Andersson, Leighton and Young120). Likewise, high levels of H2O2 reduce the activity of α-ketoglutarate dehydrogenase, one far from near-equilibrium regulatory enzyme of the TAC(Reference Tretter and Adam-vizi121). In contrast to what is observed for β-cells, reduction in FA oxidation by malonyl-CoA is not desirable for skeletal muscle fibres. Under conditions of increased ROS production, aconitase inhibition may induce citrate accumulation and cataplerosis, resulting in decreased glycolytic flux and enhanced malonyl-CoA synthesis, leading to reduced FA oxidation. In agreement with this statement, malonyl-CoA synthesis has been shown to be increased in the situations of diabetes and insulin resistance(Reference Bandyopadhyay, Yu and Ofrecio122). Acetyl-CoA carboxylase knockout mice show enhanced insulin sensitivity and were prevented from fat-rich diet-induced obesity(Reference Choi, Savage and Abu-Elheiga123).
Chronic muscle contraction (i.e. exercise) remains as the pivotal preventive therapy against peripheral insulin resistance development. Voluntary exercise stimulates blood glucose uptake, contributing to glucose homeostasis as well by a non-insulin-dependent mechanism(Reference Thorell, Hirshman and Nygren110, Reference Khayat, Patel and Klip124). In addition, exercise raises the expression of several glycolytic and oxidative enzymes involved in glucose and FA oxidation(Reference Laursen, Marsh and Jenkins125–Reference Perry, Heigenhauser and Bonen127), contributing to high exercise and resting FA oxidation by the enhancement of TAC flux(Reference Befroy, Petersen and Dufour17, Reference Holloszy, Kohrt and Hansen128, Reference Mittendorfer and Klein129). In addition to the potential effect upon oxidative enzyme activities, exercise may contribute to a high mitochondrial FA oxidation capacity by regulating TAC flux and intermediate concentrations. Exercise also improves skeletal muscle antioxidant capacity, reducing the installation of oxidative stress-induced mitochondrial impairment(Reference Ji130). During moderate- to high-intensity exercise, TAC anaplerosis is increased and has a pivotal role in maintaining muscle contraction efficiency(Reference Bowtell, Marwood and Bruce21, Reference Sahlin, Katz and Broberg22). TAC anaplerosis exerts an important role in regulating IMTG oxidation. The failure of this regulation might be implicated in peripheral insulin resistance development. In glycogen-depleted muscle, TACI concentration is maintained at the same level as during high glycogen store conditions during prolonged exercise(Reference Baldwin, Snow and Gibala131). Under muscle glycogen-depleted exercise, FA metabolism is substantially favoured, suggesting a close relationship between anaplerosis and FA oxidation. However, acute down-regulation of TACI does not reduce oxidative ATP synthesis during exercise(Reference Dawson, Baker and Greenhaff132). Therefore, the anaplerosis process may not be directly associated with oxidative ATP production in muscle skeletal cells. Further studies are required to investigate the meaning of a possible link between anaplerosis, FA oxidation control and insulin resistance in skeletal muscle.
Protein malnutrition and peripheral insulin resistance
Protein-deficient diets have been shown to markedly affect skeletal muscle function. Several reports indicate that undernourished rats present low muscle weight and impaired morphological, metabolic and functional developments(Reference Alves, Dâmaso and Dal Pai133–Reference Oumi, Miyoshi and Yamamoto135). Moreover, offspring from protein-malnourished dams also shows altered muscle structure and function that can affect animal posture and locomotion(Reference Toscano, Manhães-de-Castro and Canon35). Lehnert et al. (Reference Lehnert, Byrne and Reverter136), using complementary DNA microarrays, showed a 2- to 6-fold reduction in the expression of several muscle structural proteins and metabolic enzymes in cattle submitted to severe undernutrition. Suppressed muscle growth in undernourished rats occurs probably due to a reduction in muscle and blood insulin-like growth factor-I content(Reference Yamaguchi, Fujikawa and Tateoka137). Based on the aforementioned, one would expect that a protein deficiency-induced decrease of muscle size and function would result in reduced muscle glucose and FA metabolism and predisposing to obesity and type 2 diabetes(Reference Stannard and Johnson138, Reference Zambrano, Martínez-Samayoa and Bautista139).
Protein malnutrition shows diverse effects during the lifespan concerning peripheral insulin resistance development. Short-term protein undernutrition effects have been previously demonstrated to enhance muscle insulin sensitivity(Reference Escriva, Kergoat and Bailbé140). This effect has been shown to occur by improvement in several steps of the insulin cascade, such as increased p38 mitogen-activated protein kinase-induced GLUT-4 translocation, high levels of insulin receptor and IRS-1 tyrosine phosphorylation as well as increased IRS-1–PI3 kinase p85 subunit association and reduced IRS-1 serine phosphorylation(Reference Reis, Carneiro and Mello141–Reference Gavete, Martín and Alvarez143).
Conversely, long-term protein undernutrition has the opposite effect, mainly when higher amounts of the nutrient become available(Reference Hales and Barker144). Adult rats submitted to protein restriction in early life, or offspring from undernourished mothers, develop hyperinsulinaemia, peripheral insulin resistance and type 2 diabetes(Reference Huang, Qiu and Shen27, Reference Kappeler, De Magalhaes Filho and Leneuve145–Reference Fernandez-Twinn, Wayman and Ekizoglou147). These alterations have been demonstrated to affect the second generation(Reference Pinheiro, Salvucci and Aguila148). Indeed, evidence points to a positive correlation between reduced fetal growth, a well-known protein-restriction feature, and peripheral insulin resistance development(Reference Ozanne and Hales34, Reference Stocker, Arch and Cawthorne149, Reference Newsome, Shiell and Fall150). In contrast to short-term effects, insulin signalling seems to be compromised in the adult stage of early protein undernourishment. Reduced protein kinase C-ζ expression has been reported in rats and young men. In addition, skeletal muscle PI3 kinase–Akt insulin signalling steps have been demonstrated to be altered in low-birth-weight men and rats(Reference Fernandez-Twinn, Wayman and Ekizoglou147, Reference Jensen, Martin-Gronert and Storgaard151, Reference Ozanne, Olsen and Hansen152).
Early protein restriction has been reported to programme the appetite in late adult life(Reference Bellinger and Langley-Evans153), reducing serotonin inhibitory action on food intake, and stimulating the preference for fat-rich foods(Reference Lopes de Souza, Orozco-Solis and Grit154, Reference Bellinger, Lilley and Langley-Evans155). Furthermore, early protein undernutrition has also been shown to determine fat distribution and reduce physical activity levels, contributing to body and skeletal muscle fat storage in adult life(Reference Bellinger, Sculley and Langley-Evans156). The observed differences upon skeletal muscle glucose metabolism after early protein restriction might be associated with time- and diet-dependent metabolic changes. However, data on the metabolic regulation of protein malnutrition-induced peripheral insulin resistance are scant. To date, most studies have focused on neuroendocrine aspects and some have attempted to investigate skeletal muscle insulin signalling pathway changes. The effects of IMTG stores, incomplete FA oxidation and accumulation of metabolites leading to serine kinase activation and mitochondrial function are still unclear.
Short- and long-term protein malnutrition, anaplerosis and peripheral insulin resistance
Concerning metabolic aspects of short-term protein malnutrition, Fagundes et al. (Reference Fagundes, Moura and Passos157) reported that young adult rats from low-protein-fed mothers showed low visceral and total body fat content, probably caused by increased lipolysis or decreased lipogenesis which might be related to the observed high plasma catecholamine and low insulin levels compared with control. Pups from low-protein-fed mothers showed reduced serum NEFA and similar skeletal muscle acetyl-CoA carboxylase, FA synthase and carnitine palmitoyl transferase-1 expression as compared with control(Reference Maloney, Gosby and Phuyal158). These results are in line with those reported by Toyoshima et al. (Reference Toyoshima, Ohne and Takahashi142). These latter authors reported decreased levels of IRS-1 serine phosphorylation. The lower level of total body and consequently skeletal muscle fat might reduce the activation of specific serine kinases that impair skeletal muscle insulin downstream events. On the other hand, Gosby et al. (Reference Gosby, Stanton and Maloney159) provided clues for long-term protein restriction to increase body fat content triggering peripheral insulin resistance events. Zhu et al. (Reference Zhu, Ford and Means160) examined the long-term effects of undernutrition on skeletal muscle of offspring pregnant ewes and reported severe alterations in skeletal muscle metabolism. Despite an enhanced proportion of type 2 muscle fibres, GLUT-4 concentration was decreased and IMTG content was increased. In addition, a reduction in FA oxidation due to an almost 25 % decrease in carnitine palmitoyl transferase-1 activity, as well as a reduced expression of ATP synthase and antioxidant enzymes, probably by decreased mitochondria density, was reported. In agreement, Park et al. (Reference Park, Kim and Kim161) showed reduced mitochondrial DNA content and cytochrome c oxidase subunits I and III expression due to long-term effect of protein malnutrition during gestation and lactation.
Selak et al. (Reference Selak, Storey and Peterside162), using the bilateral uterine artery ligation model in pregnant rats to induce intra-uterine growth retardation, reported a decrease of 43 % in muscle glycogen content compared with control. This effect was associated with decreased mitochondrial ATP synthesis and reduced pyruvate, α-ketoglutarate, glutamate and succinate oxidation in isolated muscle mitochondria during respiratory state 3. Interestingly, alterations in most of the respiratory chain-linked electron transfer and energy coupling in muscle mitochondria parameters between control and growth-retarded animals were not observed. In this sense, a reduction in TAC flux and FA oxidation would be expected, but unfortunately TACI content was not measured. Evidence for a reduced TAC flux was provided by Lane et al. (Reference Lane, Chandorkar and Flozak163). These authors showed a decreased NAD+:NADH in growth-retarded rats, despite unaltered activities of mitochondrial isocitrate dehydrogenase and malate dehydrogenase(Reference Lane, Kelley and Ritov164). As discussed earlier, under nutrient deprivation, TAC flux and consequently mitochondrial metabolism are altered. Mehta et al. (Reference Mehta, Chopra and Mehta165) demonstrated reduced oxidative enzyme activities in growing young monkeys submitted to a low energy intake, being associated with peripheral insulin resistance and type 2 diabetes. However, as proposed for ordinary nutrition conditions, TAC flux might be reduced by impaired specific TAC enzyme activities or anaplerosis capacity, and so further studies are needed to answer the remaining questions.
Elevated oxidative stress has also been reported after protein malnutrition in skeletal muscle. Despite conflicting results, antioxidant enzyme activities vary according to diet protein content, and lipid peroxidation was directly related to their detoxifying capacity(Reference Park, Kim and Kim161, Reference Huang and Fwu166). Moreover, a twofold reduction in glutathione-S-transferase expression and glutathione content was observed in skeletal muscle from malnourished rats(Reference Zhu, Ford and Means160). These effects were observed to be in concert with reduced GPX scavenger capacity(Reference Oumi, Miyoshi and Yamamoto167).
The long-term metabolic effects of protein undernutrition seem, therefore, to reproduce the effects of obesity in regular-fed subjects and it is reasonable that peripheral insulin resistance and type 2 diabetes in regular feeding and protein undernutrition are induced by the same metabolic alterations. However, earlier, chronic and acute exercise is a powerful tool against peripheral insulin resistance, by its action upon restoration of insulin sensitivity. Nevertheless, early protein undernutrition seems to compromise exercise tolerance in men and rats(Reference Toscano, Manhães-de-Castro and Canon35, Reference Bellinger, Sculley and Langley-Evans156, Reference Kopple, Storer and Casburi168).
During adult life, reduced exercise capacity after early protein undernourishment was demonstrated to be associated with reduced intracellular levels of phosphocreatine and inorganic phosphate(Reference Thompson, Damyanovich and Madapallimattam169). This effect was accompanied by a faster depletion of muscle glycogen stores during exercise(Reference de-Mello170).
Conclusion
β-Cell elevated TACI concentration plays a key role for oxidative energy production and insulin release. The mechanism is supported by pyruvate metabolism (anaplerosis), mainly involving the enzyme PC. The citrate, malate and isocitrate produced exit the mitochondria to the cytosol (cataplerosis), increasing the cytosolic pyruvate concentration followed by elevated NADPH generation. Protein-deficient diets lead to impaired insulin secretion which is related to the lower mitochondrial oxidative and anaplerotic capacity. It seems reasonable, therefore, that in the protein-deficient state anaplerosis might be compromised, leading to reduced NADH:NAD+ and NADPH:NADP+ ratios, and thus further investigation is needed. In peripheral tissues, the mismatch between IMTG hydrolysis and mitochondrial β-oxidation increases the intracellular lipid content with detrimental effects on insulin signalling and glucose metabolism in normally fed and protein malnutrition states. In contrast to β-cells, skeletal muscle fibre TAC flux enhancement does not seem to be related to ATP:ADP increase, but with FA oxidation regulation. Despite no clear evidence of impaired anaplerotic and cataplerotic reactions being available in protein undernourishment, it is possible that peripheral insulin resistance could be triggered by reduced anaplerotic replenishment of the TACI, leading to inadequate FA oxidation. In addition, impaired TAC could also be induced by oxidative stress, followed by citrate cataplerosis and consequent malonyl-CoA accumulation. Therefore, based on the aforementioned data, future studies are needed to better focus on the role played by anaplerosis and cataplerosis reactions upon the control of β-cell insulin release mechanisms and peripheral insulin resistance in protein malnutrition states.
Acknowledgements
Fundacao de Amparo a Pesquisa do Estado de Sao Paulo (FAPESP), Coordenacao de Aperfeicoamento de Pessoal de Nivel Superior (CAPES) and Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPq) are acknowledged.
The authors are supported by FAPESP, CNPq and CAPES.
All authors contributed to manuscript conception, drafting and critical revision.
The authors disclose here no conflict of interest.




