


Hepcidin, a liver-secreted antimicrobial peptide, plays a central role in the control of systemic Fe homeostasis by inhibiting Fe absorption and reducing reticuloendothelial Fe sequestration. Hepatic hepcidin expression is regulated by several stimuli including body Fe status(Reference Pigeon, Ilyin and Courselaud1). The Fe-sensing pathway that regulates hepcidin involves several molecules including HFE high-Fe or haemochromatosis protein, haemojuvelin (HFE2) and transferrin receptor 2(Reference Goswami and Andrews2, Reference Lin, Goldberg and Ganz3). Defects in this Fe-sensing pathway or hepcidin itself result in hereditary haemochromatosis(Reference Feder, Gnirke and Thomas4–Reference Camaschella, Roetto and Cali7). The role of hepcidin in Fe homeostasis was revealed in mice lacking the transcription factor upstream stimulatory factor 2 (Usf2)(Reference Nicolas, Bennoun and Devaux8), which was later proposed to be one of the cis-regulatory elements of hepcidin expression(Reference Bayele, McArdle and Srai9). In this mouse model, Usf2 disruption was associated with hepcidin deficiency and subsequent severe tissue Fe overload(Reference Nicolas, Bennoun and Devaux8). However, genetic disruption of Usf2 has also been reported to affect glucose-dependent gene expression in the liver(Reference Vallet, Henrion and Bucchini10).
Circulatory Fe is acquired from dietary Fe and senescent erythrocytes through the Fe exporter ferroportin expressed in enterocytes and splenic macrophages. Hepcidin has been demonstrated to induce internalisation and degradation of ferroportin in several in vitro models including bone marrow-derived macrophages, the mouse monocyte–macrophage cell line J774, the rat cardiomyocyte cell line H9C2 and in cell lines ectopically overexpressing green fluorescent protein (GFP)-tagged ferroportin(Reference Delaby, Pilard and Goncalves11–Reference Nemeth, Tuttle and Powelson13). Some in vivo studies have indirectly suggested that this mechanism is important in tissue Fe metabolism. For example, increased ferroportin protein expression in the duodenum, liver and spleen has been shown in several hepcidin-deficient mouse models such as Hfe2 − / − and Usf2 − / − mice(Reference Huang, Pinkus and Pinkus14, Reference Viatte, Lesbordes-Brion and Lou15). An inverse relationship between the hepcidin and duodenal expression of ferroportin and divalent metal transporter 1 (DMT1) protein has also been found in pregnant and Fe-deficient rats(Reference Millard, Frazer and Wilkins16, Reference Frazer, Wilkins and Becker17). In contrast, however, hepcidin expression was suppressed in phenylhydrazine-treated rats and was associated with increased DMT1 protein in the duodenum without a change in ferroportin(Reference Frazer, Inglis and Wilkins18). In more direct studies, treatment with synthetic hepcidin and hepcidin-containing conditioned medium reduced DMT1 expression at both RNA and protein levels as well as apical Fe uptake in intestinal cell lines (Caco-2, HuTu80 and HT29) and rat duodenal segments without affecting basolateral Fe transport or ferroportin expression(Reference Yamaji, Sharp and Ramesh19–Reference Chaston, Chung and Mascarenhas21). In parallel studies, it has been shown that hepcidin caused a dramatic reduction in ferroportin expression in the THP-1 macrophage cell model (human acute monocytic leukaemia cell line). In agreement with these latter in vitro studies, hepcidin treatment in mice suppressed intestinal Fe uptake but had no effect on relative Fe transfer to blood. A significant reduction in total mucosal uptake (TMU) upon multiple hepcidin injections has been reported in CD1 mice (feeding both control and an Fe-deficient diet) as well as in Hfe wild-type/knockout mice(Reference Laftah, Ramesh and Simpson22). Interestingly, recent studies have shown that hepcidin inhibited Fe transport and enhanced cellular Fe accumulation in Caco-2 cells, suggesting a blockage of Fe efflux albeit with unaltered ferroportin expression(Reference Chung, Chaston and Marks23). Furthermore, hepcidin administration in C57BL/6 mice resulted in decreased splenic ferroportin expression within 4 h. In contrast, reduced in vivo TMU and duodenal ferroportin expression occurred at 24 h after injection. A cell type-specific response or differential tissue sensitivity to hepcidin was therefore postulated.
In previous studies, Fe absorption was measured at 24 or 72 h after hepcidin injection. However, the effects of hepcidin could be more rapid, and the findings at the 24 h time point could be secondary effects(Reference Laftah, Ramesh and Simpson22, Reference Chung, Chaston and Marks23). In addition, another key variable in such in vivo experiments is the endogenous production of hepcidin by mice, which could interfere with the effects of exogenous hepcidin. So far, direct in vivo effects of hepcidin have been studied only in wild-type (CD1 and C57BL/6) and Hfe − / − mice that express endogenous hepcidin. The present study was therefore conducted in a mouse model lacking endogenous hepcidin, namely hepcidin1 knockout (Hepc1 − / − ) mice(Reference Lesbordes-Brion, Viatte and Bennoun24), in order to explore the effects of total hepcidin deficiency. Additionally, the effects of hepcidin administration on Fe parameters, Fe absorption and ferroportin expression were also studied at an earlier time point (4 h post-injection) than previously reported.
Experimental methods
Animal care and hepcidin treatment
The generation and breeding of Hepc1 − / − mice (a mixed C57BL/6 × 129 background strain backcrossed for at least five generations on C57BL/6) has previously been reported(Reference Lesbordes-Brion, Viatte and Bennoun24). The previous study has shown no differences in Fe metabolism parameters between Hepc1 wild-type and heterozygous mice(Reference Lesbordes-Brion, Viatte and Bennoun24); hence, heterozygous mice were used as controls for the present study. Male Hepc1 − / − mice and heterozygous littermates aged 9–12 weeks old were intraperitoneally injected with 10 μg of synthetic human hepcidin (Peptide International, Louisville, KY, USA) or an equal volume of sterile saline solution. An in vivo Fe absorption study and sample collection were conducted at 4 h after the injection. Dietary Fe deficiency was induced by feeding mice a low Fe-purified diet (TD.80 396, 4 parts per million Fe; Harlan Teklad, Madison, WI, USA) for 2 weeks. In other experiments, mice were fed on the rat and mouse no. 1 maintenance (RM1) diet (Special Diets Services, Essex, UK). The dietary composition of the RM1 diet is supplied by the manufacturer as follows: metabolisable energy 10·74 kJ/g, oil 7·42 % energy, protein 17·49 % energy, carbohydrate 75·09 % energy, moisture 10 %, minerals 33·29 g/kg (Fe 159·30 parts per million) and vitamins 3·64 g/kg. Mice were fed ad libitum on diets up until the time of culling. The mice were culled under isoflurane-induced anaesthesia by exsanguination. National guidelines for the care and use of animals were followed. All experimental procedures involving animals were approved by the UK Home Office.
In vivo iron absorption
In vivo Fe absorption was measured in tied-off duodenal segments as described previously(Reference Laftah, Ramesh and Simpson22). In brief, the experiments were conducted in anaesthetised mice. A duodenal segment was tied at both ends followed by the injection of 250 μm-59FeNTA2 (ferric nitrilotriacetate) in physiological buffer (125 mm-NaCl, 3·5 mm-KCl, 10 mm-MgCl2, 1 mm-CaCl2 and 16 mm-HEPES, pH 7·4) into the tied-off segment (radiolabelled Fe was obtained as 59FeCl3; Perkin Elmer, Boston, MA, USA). The segment was placed back into the abdominal cavity. After 10 min incubation, the duodenal segment was flushed with ice-cold saline solution and weighed. Blood, liver and spleen were collected. Radioactivity in tissue samples and blood was measured using a gamma counter (1282 Compugamma; LKB Wallac, Turku, Finland), while carcasses were counted for radioactivity by a high-resolution bulk sample counter (J&P Engineering, Reading, UK). Radioactivity in the duodenum is referred to as mucosal retention (MR), while radioactivity in carcass and other tissues is regarded as mucosal transfer (MT). TMU is the amount of total radioactive Fe absorbed from the gut lumen, and the percentage of MT (%MT) is the relative amount of Fe transfer into the body in comparison with total Fe uptake.
Hepcidin quantitative RT-PCR
RNA was extracted from the liver using TRIzol reagent (Invitrogen, Paisley, UK), and complementary DNA was synthesised using a Transcriptor High Fidelity cDNA Kit (Roche Diagnostics, Mannheim, Germany). Quantitative RT-PCR was performed using the ABI Prism 7000 (Applied Biosystems, Carlsbad, CA, USA) and Universal ProbeLibrary System (Roche Diagnostics). Hepcidin (Hepc1) expression was normalised to β-actin (Actb) mRNA. The sequence of the utilised primers is listed as follows:
ActbForward: CTAAGGCCAACCGTGAAAAG
Reverse: ACCAGAGGCATACAGGGACA
Hepc1Forward: GATGGCACTCAGCACTCG
Reverse: GCTGCAGCTCTGTAGTCTGTCT
Measurement of Hb, serum iron and tissue non-haem iron contents
Hb was determined spectrophotometrically using the methods described by Beutler(Reference Beutler25). Serum Fe was measured with a liquid ferrozine-based Fe reagent (Thermo Electron, Melbourne, VIC, Australia). Tissue non-haem Fe levels were determined by a modification of the method of Foy et al. (Reference Foy, Williams and Cortell26), as described by Simpson & Peters(Reference Simpson and Peters27).
Western blot analysis of ferroportin
Membrane protein was extracted by homogenising tissues in 1 ml of lysis buffer (20 mm-KH2PO4, 0·1 mm-EDTA and 135 mm-KCl) and protease inhibitor cocktail (1:200 dilution; Sigma-Aldrich, Poole, UK). The homogenate was centrifuged at 1000 g for 5 min at 4°C. The resulting supernatant was decanted to new tubes and centrifuged at 28 500 g for 1 h at 4°C. The supernatant was discarded, and the pellet was resuspended in a buffer containing 20 mm-KH2PO4, 10 mm-EDTA, 1 mm-KCl and protease inhibitor cocktail. Western blot analysis was performed using a primary rabbit anti-mouse metal transporter protein 1 (MTP1) antibody (Alpha Diagnostic, San Antonio, TX, USA) and rabbit anti-actin antibody (Sigma-Aldrich) in order to detect ferroportin and actin, respectively.
Ferroportin immunofluorescence
Tissue samples were immersed in tissue-freezing medium (TBS, Durham, NC, USA). The tissue blocks were cut into 5 μm sections and mounted on poly-lysine-coated slides (Sigma-Aldrich). The slides were air-dried and fixed in acetone. Ferroportin immunofluorescence was conducted using rabbit anti-mouse ferroportin antiserum (1:100 dilution)(Reference McKie, Marciani and Rolfs28) and fluorescein isothiocyanate-conjugated anti-rabbit antibody (Dako Cytomation, Glostrup, Denmark). The images were captured with a DM IRE2 confocal microscope (Leica, Wetzlar, Germany).
Statistical analysis
Data are presented as means with their standard errors. The comparison of multiple groups for significant effects of two variables (hepcidin injection and genotype) was determined by two-way ANOVA with a Bonferroni post hoc test. Relative hepcidin mRNA expression was analysed by Student's unpaired t test. A P value less than 0·05 was considered as significant. All statistical analyses were performed using GraphPad Prism 4 software (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Hepcidin inhibited iron absorption in Hepc1−/− mice
In order to assess the influence of hepcidin on intestinal Fe absorption, radiolabelled Fe uptake was performed in vivo using tied-off duodenal segments from anaesthetised mice fed a standard laboratory diet.
In comparison with heterozygous littermates, Hepc1 − / − mice had about a two- to threefold increase in TMU, MR and MT (P < 0·001 for the effects of genotype on TMU and MT; P = 0·001 for the effect of genotype on MR), whereas no significant difference was found in %MT (Table 1). Hepcidin administration reduced TMU and MT in both genotypes but with significant reductions in Hepc1 − / − mice 4 h after the treatment (P = 0·003 and 0·001 for the effects of hepcidin on TMU and MT, respectively). Interestingly, a borderline significant effect of hepcidin treatment on %MT is indicated by two-way ANOVA (Table 1).
Table 1 Effects of hepcidin on iron parameters and iron absorption in mice fed a standard laboratory diet
(Mean values with their standard errors)

Het, heterozygote; KO, Hepc1 − / − mice; TMU, total mucosal uptake; MR, mucosal retention; MT, mucosal transfer; %MT, percentage of mucosal transfer.
* Mean value was significantly different from that of the heterozygote control (P < 0·05).Mean values were significantly different from those of the Hepc1 − / − control: ††P < 0·01, †††P < 0·001.
Tissue distribution of the absorbed 59Fe was also compared between hepcidin-injected Hepc1 − / − and heterozygous mice. Hepc1 − / − mice demonstrated significantly higher radioactivity in the liver, while less 59Fe was found in the spleen and erythrocytes compared with heterozygous littermates (P < 0·001 for the effects of genotype on radioactivity in the liver; P = 0·002 and 0·030 for the effects of genotype on radioactivity in the spleen and erythrocytes, respectively; Table 2).
Table 2 Distribution of gastrointestinally absorbed 59Fe in mice fed a standard laboratory diet*
(Mean values with their standard errors)
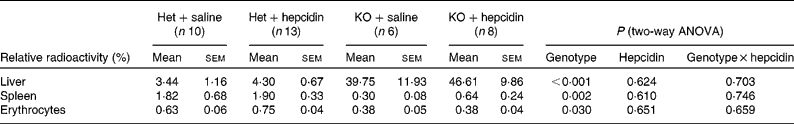
Het, heterozygote; KO, Hepc1 − / − mice.
* Radioactivity in the samples obtained from in vivo Fe absorption is presented relative to the total counts in the body apart from the duodenum. The relative radioactivity in erythrocytes was normalised to the relative radioactivity of the whole blood samples. Statistical analysis was performed by two-way ANOVA with a Bonferroni post hoc test.
Hepcidin administration did not reduce serum iron in Hepc1−/− mice
Serum and tissue Fe levels were also measured to determine the effect of hepcidin on Fe compartmentalisation. As expected, Hepc1 − / − mice contained significantly higher serum Fe and liver non-haem Fe levels compared with heterozygous littermates (P < 0·001 for the effects of genotype on serum Fe and liver non-haem Fe levels; Table 1). Notably, hepcidin treatment resulted in a 37 % reduction of serum Fe in heterozygotes (33·3 (sem 1·8) v. 21·0 (sem 3·7) μm, P < 0·05 by the Bonferroni post hoc test; Table 1), while no change in serum Fe levels was found in Hepc1 − / − mice.
Hepcidin injection suppressed endogenous hepcidin expression in heterozygous mice and ferroportin expression in Hepc1−/− mice
Quantitative RT-PCR suggested a significant reduction in liver hepcidin expression in heterozygous mice upon exogenous hepcidin treatment (Fig. 1).

Fig. 1 Effect of exogenous hepcidin treatment on endogenous hepcidin expression in Hepc1 heterozygous mice. Quantitative RT-PCR of hepcidin mRNA (Hepc1) from the liver of saline (![]() )- and hepcidin (
)- and hepcidin (![]() )-treated Hepc1 heterozygous mice. Relative expression was acquired by normalising Hepc1 mRNA to β-actin (Actb) mRNA. Values are means, with standard errors represented by vertical bars, for the fold change as compared with the saline-treated group (n 4). The samples were measured in triplicate. Statistical analysis was performed by Student's unpaired t test. ** Mean value was significantly different from that of the heterozygote control (P < 0·01).
)-treated Hepc1 heterozygous mice. Relative expression was acquired by normalising Hepc1 mRNA to β-actin (Actb) mRNA. Values are means, with standard errors represented by vertical bars, for the fold change as compared with the saline-treated group (n 4). The samples were measured in triplicate. Statistical analysis was performed by Student's unpaired t test. ** Mean value was significantly different from that of the heterozygote control (P < 0·01).
Western blot analysis showed higher ferroportin protein expression in the spleen and liver along with a trend towards increased ferroportin expression in the duodenum of Hepc1 − / − mice compared with heterozygous mice (P = 0·109, P < 0·001 and P = 0·002 for the effects of genotype on ferroportin expression in the duodenum, liver and spleen, respectively; Figs. 2–4). Intraperitoneal hepcidin injection in the knockout mice significantly reduced ferroportin protein expression in the spleen (P < 0·05 by the Bonferroni post hoc test; Fig. 4). No change in ferroportin expression upon hepcidin administration was found in the heterozygotes.

Fig. 2 Effect of hepcidin administration on duodenal ferroportin expression in heterozygous and Hepc1 − / − mice. (a) Western blot analysis of ferroportin (50 μg crude membrane preparation) from the duodenum of saline (![]() )-/hepcidin (
)-/hepcidin (![]() )-injected heterozygous and Hepc1 − / − mice. Molecular weight markers are indicated on the left. (b) Blot densitometry as obtained by ImageJ software (National Institutes of Health, Bethesda, MD, USA). Ferroportin expression was normalised to the expression of β-actin and presented in arbitrary unit (AU). Values are means, with standard errors represented by vertical bars (n 5, 4, 5 and 4, respectively). Statistical analysis was performed by two-way ANOVA with a Bonferroni post hoc test. No significant effects of genotype, hepcidin and genotype × hepcidin: P = 0·109, 0·207, 0·560, respectively.
)-injected heterozygous and Hepc1 − / − mice. Molecular weight markers are indicated on the left. (b) Blot densitometry as obtained by ImageJ software (National Institutes of Health, Bethesda, MD, USA). Ferroportin expression was normalised to the expression of β-actin and presented in arbitrary unit (AU). Values are means, with standard errors represented by vertical bars (n 5, 4, 5 and 4, respectively). Statistical analysis was performed by two-way ANOVA with a Bonferroni post hoc test. No significant effects of genotype, hepcidin and genotype × hepcidin: P = 0·109, 0·207, 0·560, respectively.

Fig. 3 Effect of hepcidin administration on ferroportin expression in the liver of heterozygous and Hepc1 − / − mice. (a) Western blot analysis of ferroportin (100 μg crude membrane preparation) from the liver of saline (![]() )-/hepcidin (
)-/hepcidin (![]() )-injected heterozygous and Hepc1 − / − mice. Molecular weight markers are indicated on the left. (b) Blot densitometry as obtained by ImageJ software (National Institutes of Health, Bethesda, MD, USA). Ferroportin expression was normalised to the expression of β-actin and presented in arbitrary unit (AU). Values are means, with standard errors represented by vertical bars (n 5, 4, 5 and 4, respectively). Statistical analysis was performed by two-way ANOVA with Bonferroni post hoc test. Mean values were significant for the effect of genotype: P < 0·001. No significant effects of hepcidin and genotype × hepcidin: P = 0·301 and 0·824, respectively.
)-injected heterozygous and Hepc1 − / − mice. Molecular weight markers are indicated on the left. (b) Blot densitometry as obtained by ImageJ software (National Institutes of Health, Bethesda, MD, USA). Ferroportin expression was normalised to the expression of β-actin and presented in arbitrary unit (AU). Values are means, with standard errors represented by vertical bars (n 5, 4, 5 and 4, respectively). Statistical analysis was performed by two-way ANOVA with Bonferroni post hoc test. Mean values were significant for the effect of genotype: P < 0·001. No significant effects of hepcidin and genotype × hepcidin: P = 0·301 and 0·824, respectively.

Fig. 4 Effect of hepcidin administration on ferroportin expression in the spleen of heterozygous and Hepc1 − / − mice. (a) Western blot analysis of ferroportin (50 μg crude membrane preparation) from the spleen of saline (![]() )-/hepcidin (
)-/hepcidin (![]() )-injected heterozygous and Hepc1 − / − mice. Molecular weight markers are indicated on the left. (b) Blot densitometry as obtained by ImageJ software (National Institutes of Health, Bethesda, MD, USA). Ferroportin expression was normalised to the expression of β-actin and presented in arbitrary unit (AU). Values are means, with standard errors represented by vertical bars (n 5, 4, 5 and 4, respectively). Statistical analysis was performed by two-way ANOVA with a Bonferroni post hoc test. Mean values were significant for the effect of genotype and hepcidin: P = 0·002, 0·037, respectively. No significant effect of genotype × hepcidin: P = 0·074. * Mean value was significantly different as from that of the knockout control (P < 0·05).
)-injected heterozygous and Hepc1 − / − mice. Molecular weight markers are indicated on the left. (b) Blot densitometry as obtained by ImageJ software (National Institutes of Health, Bethesda, MD, USA). Ferroportin expression was normalised to the expression of β-actin and presented in arbitrary unit (AU). Values are means, with standard errors represented by vertical bars (n 5, 4, 5 and 4, respectively). Statistical analysis was performed by two-way ANOVA with a Bonferroni post hoc test. Mean values were significant for the effect of genotype and hepcidin: P = 0·002, 0·037, respectively. No significant effect of genotype × hepcidin: P = 0·074. * Mean value was significantly different as from that of the knockout control (P < 0·05).
The effect of hepcidin on ferroportin was further investigated by immunofluorescence. Ferroportin staining in the duodenum, liver and spleen was stronger in Hepc1 − / − mice than in heterozygotes (Fig. 5). Upon hepcidin administration, ferroportin expression in all three tissues of the treated knockout mice was reduced in comparison with control Hepc1 − / − mice. However, the difference between the control and treatment groups was not distinct in heterozygous mice. Similar to Usf2 − / − mice(Reference Viatte, Lesbordes-Brion and Lou15), ferroportin immunofluorescence in the Hepc1 − / − liver was detected predominantly in Kupffer cells.

Fig. 5 Immunofluorescence study of the effect of hepcidin administration on ferroportin expression in heterozygous and Hepc1 − / − mice. The expression and localisation of ferroportin in the duodenum, liver and spleen of saline- or hepcidin-injected heterozygous/Hepc1 − / − mice was demonstrated through the immunofluorescence technique. Ferroportin protein was visualised as green fluorescence, and the nuclei were counterstained in red. Images were captured from a Leica DM IRE2 confocal microscope (Leica, Wetzlar, Germany). Original magnification, 400 × (top two rows), 200 × (bottom row).
Iron deficiency altered the effects of hepcidin treatment
To explore whether the effects of hepcidin depended on Fe status/dietary Fe levels, mice were fed a low-Fe diet for 2 weeks before the in vivo Fe absorption study. Under the Fe-deficient diet treatment, Hepc1 − / − mice had significantly increased serum Fe as well as liver non-haem Fe levels compared with heterozygous mice (P < 0·001 for the effects of genotype on serum Fe and liver non-haem Fe levels; Table 3). A significant effect of hepcidin on serum Fe (P = 0·016) was indicated by two-way ANOVA. However, no significant difference in serum Fe and liver non-haem Fe levels was found between the saline- and hepcidin-treated groups.
Table 3 Effects of hepcidin on iron absorption in mice fed an iron-deficient diet
(Mean values with their standard errors)

Het, heterozygote; KO, Hepc1 − / − mice; TMU, total mucosal uptake; MR, mucosal retention; MT, mucosal transfer; %MT, percentage of mucosal transfer.
Under low-Fe diet feeding, heterozygous mice generally had higher TMU than those on the RM1 diet. TMU was significantly higher, and MT was marginally increased in Hepc1 − / − compared with heterozygous mice (P = 0·044 and 0·069 for the effects of genotype on TMU and MT, respectively; Table 3). No significant difference in MR and % MT was found between heterozygous and knockout mice. Furthermore, hepcidin injection had no significant effect on any of the Fe absorption parameters in either genotype.
Discussion
Fe loading in most of hereditary haemochromatosis patients is caused by continuously enhanced Fe absorption due to reduced hepcidin levels. Fe absorption has been shown to be higher in Hfe − / − mice but only to a small extent(Reference Laftah, Ramesh and Simpson22). In the present study, Fe absorption was measured in vivo in tied-off duodenal segments of Hepc1 − / − mice and heterozygous littermates. This mouse model has previously been shown to have severe systemic Fe overload with splenic macrophage sparing, whereas no difference in haematological and Fe parameters was detected between wild-type and heterozygous mice(Reference Lesbordes-Brion, Viatte and Bennoun24). Furthermore, genetic disruption of Hepc1 had no effect on body weight until the age of 8 months when significant weight loss was noted in the knockout mice. In addition, unpublished results in our laboratory found no difference in body weight and food intake between Hepc1 − / − mice and heterozygous littermates (P. Masaratana, unpublished results).
Under standard laboratory diet feeding, genetic disruption of the Hepc1 gene resulted in increased apical Fe uptake and intestinal Fe retention. However, no change in proportional basolateral Fe transfer was found. These findings are therefore in agreement with previous observations that hepcidin deficiency is associated with enhanced intestinal absorption and subsequent Fe overload. Most of the gastrointestinally absorbed Fe was taken up by the liver, explaining the extensive liver Fe loading found in this mouse model. A similar finding was also observed in hypotransferrinaemic mice(Reference Dickinson, Devenyi and Connor29–Reference Simpson, Lombard and Raja31).
The present study demonstrates that hepcidin had rapid inhibitory effects on Fe absorption particularly in Hepc1 − / − mice. The administration of synthetic hepcidin in Hepc1 − / − mice caused reductions in mucosal Fe uptake and MT at 4 h after treatment without altering the tissue distribution of absorbed Fe. Hepc1 − / − mice may be more sensitive to hepcidin treatment than heterozygous mice, as the effects of hepcidin on Fe absorption are more pronounced in the former group. This is also supported by a significant interaction between the effect of genotype and hepcidin on MT as demonstrated by two-way ANOVA. Notably, hepcidin injection in Hepc1 − / − mice exerted the strongest effect on MT. A borderline reduction in %MT upon hepcidin treatment was also found in the knockout mice, suggesting that hepcidin may modulate the basolateral transfer of the absorbed Fe which involves ferroportin.
Upon Fe-deficient diet treatment, liver non-haem Fe levels were generally lower than the mice maintained under the standard diet across the four groups, suggesting that Fe status was affected by this dietary treatment regimen. Notably, the differences in Fe absorption between Hepc1 − / − and heterozygous mice became less apparent under Fe-deficient diet feeding conditions. The effects of genotype of TMU, MR and MT are less significant than those seen in the mice fed a normal commercial diet. It is likely that the effects of Fe deficiency in enhancing Fe absorption were not additive to Hepc1 ablation or, perhaps, Fe absorption had reached its maximum capacity by Hepc1 disruption per se. Hypoxia-inducible factor (HIF)-2α expression is reported to be induced by dietary Fe deficiency; thus, Fe deprivation can exert its effects on Fe absorption through hepcidin-independent mechanisms(Reference Shah, Matsubara and Ito32). Increased protein levels of HIF-2α were associated with large increases in the duodenal expression of DMT1 and Dcytb, with a trend towards increased duodenal ferroportin and hephaestin levels despite the lack of hepcidin response(Reference Shah, Matsubara and Ito32). In agreement with this latter study, intestinal-specific disruption of the HIF pathway counteracted the effects of Fe deficiency on the expression of DMT1 and Dcytb(Reference Mastrogiannaki, Matak and Keith33). The lack of the effects of hepcidin on Fe absorption in Hepc1 − / − mice under Fe-deficient diet feeding conditions also supports the idea that the effect of hepcidin is related to enterocyte Fe levels and/or Fe status. In contrast to the present study, previous works reported that hepcidin inhibits Fe absorption independently of dietary Fe level or Fe loading(Reference Laftah, Ramesh and Simpson22). The discrepant effects of hepcidin on Fe absorption in Fe-deficient mice between the two studies may result from several factors. Previous studies have been conducted in CD1 mice fed with an Fe-deficient diet for 3 weeks followed by two doses of 50 μg hepcidin. Fe absorption was measured at 24 h after the second hepcidin injection. The present study was performed in mice with a mixed C57BL/6 × 129 background maintained on an Fe-deficient diet for 2 weeks. In addition, only a single dose of 10 μg hepcidin was administered 4 h before the Fe absorption studies. The difference in mouse strain, hepcidin dosage and treatment duration may be responsible for the discordant effects of hepcidin on Fe absorption.
In order to understand the molecular mechanism behind the hepcidin-mediated inhibition of Fe absorption as well as the hepcidin–ferroportin relationship in hepcidin-deficient subjects, hepcidin and ferroportin expression was studied. Ferroportin immunofluorescence studies showed an inverse relationship between hepcidin and ferroportin expression in the duodenum, liver and spleen of Hepc1 − / − mice. Hepcidin treatment in Hepc1 − / − mice was associated with reduced ferroportin staining in all three tissues. In addition, the change in the staining pattern was suggestive for ferroportin internalisation, particularly in the Kupffer cells of hepcidin-injected Hepc1 − / − mice (data not shown). Furthermore, Western blot analysis revealed that hepcidin administration significantly reduced ferroportin in the spleen of the knockout mice while reductions in the duodenum and liver were marginal, which was probably due to high individual variation in ferroportin expression. These data suggest that hepcidin influenced ferroportin expression in all three tissues, with a more pronounced effect in the spleen. Hepcidin has previously been shown to inhibit ferroportin expression only in the spleen(Reference Chaston, Chung and Mascarenhas21). However, hepcidin-mediated ferroportin degradation has recently been reported in primary mouse hepatocytes(Reference Ramey, Deschemin and Durel34). It should also be noted that in vivo ferroportin expression could be influenced by several factors in addition to hepcidin. This is evident in mice with intestine-specific iron-responsive protein (IRP) 1 and IRP2 ablation, which contain higher ferroportin protein levels in the duodenum despite increased hepcidin expression(Reference Galy, Ferring-Appel and Kaden35). This information suggests that the effects of hepcidin on duodenal ferroportin expression can be overridden by IRP or transcriptional controls. The implication of this regulation remains to be elucidated. Interestingly, exogenous hepcidin injection was shown to suppress endogenous hepcidin expression in heterozygous mice. This could be a direct negative feedback control from hepcidin injection or a secondary effect of hypoferraemia induced by hepcidin treatment. Hence, endogenous hepcidin suppression might be responsible for the lack of significant effects of exogenous hepcidin on Fe absorption and splenic ferroportin expression in heterozygotes.
In addition, the injection of 10 μg hepcidin failed to reduce serum Fe in Hepc1 − / − mice despite hypoferraemia induced by the same dose of hepcidin in heterozygous littermates. This finding may indicate different ferrokinetics or tissue hepcidin sensitivity between Hepc1 − / − mice and heterozygous littermates. Notably, Western blot analysis showed that although hepcidin reduced splenic ferroportin in knockout mice, the protein levels are still higher than in heterozygous mice. This may be responsible for the lack of change in serum Fe upon hepcidin treatment in Hepc1 − / − mice. A significant effect of hepcidin on serum Fe has been found to be in agreement with findings reported by Rivera et al. (Reference Rivera, Nemeth and Gabayan36), which were obtained from similar strain mice fed a similar diet. Note that their study involved small groups of mice in mixed sex while we used larger groups of male mice only, as sex is known to affect Fe metabolism(Reference Courselaud, Troadec and Fruchon37). It should also be noted that hepcidin used in the two studies was obtained from different sources. Collectively, our data suggest that the effects of exogenous hepcidin on serum Fe and Fe absorption may be affected by Fe status and/or endogenous hepcidin levels. Further studies are required with higher hepcidin dosages in order to explore these issues.
In conclusion, the crucial role of hepcidin in the pathogenesis of hereditary Fe overload was demonstrated in a Hepc1 − / − mouse model. Hepcidin deficiency was associated with a two- to threefold increase in Fe absorption and significantly higher uptake of absorbed Fe by the liver. In addition, exogenous hepcidin administration was shown to rapidly inhibit Fe absorption especially in Hepc1 − / − mice. Reduced ferroportin expression in response to hepcidin treatment was found particularly in the spleen. Finally, the response to hepcidin treatment seems to differ between Hepc1 − / − and heterozygous mice and is not observed in mice fed a low-Fe diet, suggesting that dietary Fe levels or Fe status may influence the effects of hepcidin on Fe metabolism.
Acknowledgements
The present study was supported by a grant from the European Commission (LSHM-CT-2006-037296: EUROIRON1). P. M.'s studentship was supported by the Overseas Research Students Awards Scheme and Anandamahidol Foundation. The authors have no conflicts of interest. P. M., R. J. S. and A. T. M. designed the study; P. M. and A. H. L. conducted the study; P. M., G. O. L.-D., R. J. S. and A. T. M. analysed the data; S. V. provided Hepc1 − / − mice for the study; P. M., R. J. S. and A. T. M. wrote the manuscript; G. O. L. and S. V. revised the manuscript; A. T. M. had the primary responsibility for the final content.








