


Patients with obstructive jaundice are prone to septic complications and renal dysfunction, both of which contribute to high morbidity and mortality rates(Reference Pain, Cahill and Bailey1). The main pathological basis of obstructive jaundice is impaired intestinal barrier functioning. Most previous research has focused on bacterial translocation, Kupffer cell functioning, the inflammatory response, intestinal blood barriers and blood oxidative stress. Few studies have examined the relationship between hyperbilirubinaemia and the intestinal epithelial barrier.
Recently, Raimondi et al. reported that unconjugated bilirubin (UCB) increased the permeability of the intestinal epithelium in an in vitro model(Reference Raimondi, Crivaro and Capasso2). Further, this effect was reversible. While Caco-2 cell monolayers treated for 6 h with 50 nm-UCB showed significant occludin redistribution and decreased transepithelial electrical resistance (TER), after 48 h of treatment with UCB, these effects were reversed(Reference Raimondi, Crivaro and Capasso2). Yang et al. reported that adding bile to the culture medium of IEC-6 monolayers decreased epithelial permeability and increased epithelial expression of zonula occludens (ZO)-1 and occludin. Additionally, adding bile to the medium led to phosphorylation of the mitogen-activated protein kinase ERK1/2(3). Little is known about the molecular events that connect UCB to intestinal permeability dysfunction.
Tight junctions are the uppermost basolateral connection between neighbouring enterocytes, and they constitute an important component of the epithelial barrier(Reference Mitic, Van Itallie and Anderson4). Tight junctions are composed of transmembrane proteins such as occludins, claudins and junctional adhesion molecules (JAM). The cytosolic domains of these proteins interact with the peripheral junctional proteins of the ZO family (ZO-1, ZO-2); targets for interaction also include protein kinases, especially protein kinase C (PKC) isoforms, for regulatory purposes(Reference Schneeberger and Lynch5). Tight junction assembly and paracellular permeability are regulated by a network of signalling pathways that involve different PKC isoforms(Reference Stuart and Nigam6).
Previous studies have reported impaired intestinal barrier function, significant bacterial translocation and significantly increased intestinal permeability in clinical and experimental obstructive jaundice. Using immunohistochemistry, Assimakopoulos et al. recently demonstrated that intestinal mucosal barrier dysfunction in experimental obstructive jaundice is associated with regional loss of occludin expression in the intestinal epithelium, mainly in the upper part of the villi(Reference Assimakopoulos, Scopa and Charonis7). Perioperative symbiotic treatment contributed to the maintenance of a favourable intestinal environment during biliary cancer surgery(Reference Sugawara, Nagino and Nishio8), and live probiotic Bifidobacterium lactis bacteria have been shown to inhibit toxic effects in epithelial cell culture(Reference Lindfors, Blomqvist and Juuti-Uusitalo9). Lactobacillus plantarum (LP) inhibits epithelial barrier dysfunction and IL-8 secretion induced by TNF-α(Reference Ko, Yang and Chang10) and prevents cytokine-induced apoptosis in intestinal epithelial cells(Reference Yan and Polk11).
UDP-glucuronosyltransferases 1 family-polypeptide A1 (UGT1A1) is the main isoform responsible for the glucuronidation of UCB(Reference Ohnishi and Emi12). UGT1A1-dependent bilirubin conjugation plays a critical role in the detoxification of bilirubin. After the catalysis of UGT1A1 in liver cells, UCB is transformed into water-soluble non-toxic conjugated bilirubin. Recent studies using catalytic activity assays and Western and Northern blotting of Caco-2 human intestinal cells have demonstrated a high level of induction of UGT1A1 by the flavonoid chrysin (5,7 dihydroxyflavone)(Reference Smith, Graham and Krol13). Thus, we wondered whether UCB itself could induce UGT1A1 expression in Caco-2 cells and then be transformed into conjugated bilirubin, thereby decreasing the UCB concentration to a relatively low level and finally reducing the cytotoxicity of UCB. Probiotic bacteria have been shown to have beneficial effects on intestinal barrier function. Therefore, we also hypothesised that the probiotic bacterium LP may prevent intestinal integrity disruption by UCB.
The goals of the present study were to investigate in vitro the effects of probiotic bacteria and UCB on intestinal epithelial cell viability and to assess the morphological changes and expression of tight junction proteins. Further evaluations were performed to determine the expression of PKC and UGT1A1 proteins. These studies will help to elucidate the mechanisms of bilirubin and probiotic bacterial activity in intestinal epithelial permeability.
Materials and methods
Antibodies and reagents
Rabbit polyclonal anti-occludin, rabbit polyclonal anti-JAM-A, rabbit polyclonal anti-claudin-1, mouse monoclonal anti-claudin-4, fluorescein isothiocyanate (FITC)-conjugated secondary antibodies and rabbit polyclonal anti-phosphoserine antibodies were supplied by Zymed (Invitrogen, Carlsbad, CA, USA). Rabbit polyclonal anti-ZO-1, rabbit polyclonal anti-PKC and rabbit polyclonal anti-UGT1A1 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). FITC-phalloidin was obtained from Sigma (St Louis, MO, USA). Biotin-labelled goat anti-rabbit IgG and horseradish peroxidase (HRP)-labelled streptavidin were obtained from DAKO (Glostrup, Denmark) and cell culture chemicals were obtained from GIBCO (Invitrogen). UCB was purchased from Sigma-Aldrich (St Louis, MO, USA). For experimental use, UCB was dissolved as a 10 mmol/l stock solution in 0·1 m-NaOH. Final concentrations of UCB solutions ranged from 10 to 1000 nmol/l with pH 7·4, after adjustment with chloridric acid. Experiments with UCB were performed under dim light to prevent photodegradation. All other reagents were of analytical grade and were also purchased from Sigma.
Preparation of bacteria
An LP strain (China General Microbiological Culture Collection Center (CGMCC) no. 1258) was donated as a gift from Dr Hang Xiaomin (Institute of Science Life of Onlly, Shanghai Jiao Tong University, Shanghai, China). The bacteria were grown overnight at 37°C in de Man–Rogosa–Sharpe (MRS) broth (Difco Laboratories, Detroit, MI, USA) and were centrifuged, washed and re-suspended in cold Dulbecco's PBS. Quantification of the bacterial suspension was performed using a standard curve for visible absorbance (600 nm, Beckman DU-50 spectrophotometer; Beckman Coulter, Fullerton, CA, USA). Next, 1 × 108 colony-forming units (CFU) of LP per ml were added to the Caco-2 cells. Untreated cells were used as controls in all experiments.
Cell culture
Human Caco-2 intestinal cells were purchased from the Cell Institute Affiliated Chinese Science Research Institute (Shanghai, China). The cells were plated at a density of 2 × 105 per 25 cm2 cell culture flask (Corning Inc., Corning, NY, USA). Cells were grown in Dulbecco's modified Eagle medium containing 25 mm-glucose and supplemented with 10 % fetal bovine serum, 1 % non-essential amino acids, 2 mm-l-glutamine, 1 % penicillin–streptomycin and 1 % sodium pyruvate. Cells were maintained in a humidified atmosphere of 5 % CO2 at 37°C. Caco-2 cells were cultured for 14 d before experiments. Under experimental conditions, conventional media were replaced by phenol red-and serum-free medium to avoid additional protein binding to bilirubin and interference with fluorescent tracer measurements. UCB, LP or experimental medium was added to the apical (luminal) side of the monolayer.
Experimental design
Raimondi et al. (Reference Raimondi, Crivaro and Capasso2) reported that UCB (50 nmol/l) increased the permeability of intestinal epithelia in an in vitro model and that this effect was reversible. Furthermore, they showed that a UCB concentration of 600 nmol/l was significantly cytotoxic. Therefore, the present study was designed as follows. Caco-2 cell monolayers were divided into four groups: group A, the UCB group, treated with UCB (50 nmol/l) for 6 h; group B, the LP before UCB group, treated with LP (108 CFU/l) for 1 h, then the LP was discarded and the cells were treated with UCB (50 nmol/l) for 6 h; group C, the LP following UCB group, treated with UCB (50 nmol/l) and LP (108 CFU/l) for 1 h, then the LP was discarded and the cells were treated with UCB (50 nmol/l) for another 5 h; group D, the control group. To study the UGT1A1 expression, the incubation time with UCB (50 nmol/l) was prolonged to 24 h. For the apoptosis assays, the UCB concentration was changed to 1000 nmol/l, but the UCB incubation time remained at 6 h. All experiments were conducted according to the guidelines set forth by the Ethics Committee of Shanghai Sixth People's Hospital, Shanghai, China.
Cytotoxicity assays
Caco-2 cell monolayers were exposed for 6 h to increasing concentrations of UCB (from 10 to 1000 nm), and cytotoxicity was evaluated by 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2, 4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-8) colorimetric assay. Caco-2 cells (5000 cells per well) were seeded into ninety-six-well cell plates (Corning Inc.) in 100 μl serum-free Dulbecco's modified Eagle medium cell culture medium (Gibco BRL, Grand Island, NY, USA) for 24 h before drug exposure. After 24 h of pre-incubation, cells were treated with various concentrations of UCB. After drug exposure, the media were discarded and replaced with 90 μl fresh media, followed by addition of 10 μl WST-8 solution (Cell Counting Kit; Dojindo Laboratories, Kumamoto, Japan), and incubated for 4 h at 37°C in an incubator. Cell viability was determined via colorimetric comparison by reading optical density (OD) values from a microplate reader (SoftMax, Molecular Devices, Sunnyvale, CA, USA) at an absorption wavelength of 450 nm. The percentage of surviving cells was calculated as ((experimental well − blank well)/(untreated control well − blank well)) × 100 %.
Assessment of Caco-2 apoptosis using flow cytometry
Caco-2 cell monolayers were divided into four groups: A, UCB (1000 nmol/l) only; B, LP (108 CFU/l) before UCB (1000 nmol/l); C, LP (108 CFU/l) after UCB (1000 nmol/l); D, control. After the appropriate experimental treatment, Caco-2 cells were trypsinised and assessed for apoptosis using the annexin V–FITC apoptosis detection kit (Beckman Coulter, Fullerton, CA, USA). Annexin V–FITC was used to stain apoptotic cells, and propidium iodide was used to stain necrotic cells. Cells were harvested and suspended in binding buffer at 5 × 105–1 × 106 cells/ml. The cell suspension was mixed with FITC-labelled annexin V. Cells were re-suspended in binding buffer after being rinsed with PBS buffer, and propidium iodide (final concentration 1 μg/ml) was added to the reaction. This procedure was conducted in a relatively light-free environment. Apoptotic cells were quantitatively determined using a FACScan flow cytometer (Beckman Coulter, ELITE, Fullerton, CA, USA).
Transepithelial electrical resistance experiments
Monolayers of Caco-2 cells were grown on filters (Millicell culture plate inserts; 0·4 μm pore size; 0·6 cm2). At full confluence (15–18 d), monolayers with a basal reading between 450 and 500 Ω/cm2 were used for the study and measured using a voltmeter (Millicell-ERS; Millipore, Bedford, MA, USA). The integrity of the confluent polarised monolayers was checked by measuring TER at different time intervals after treatment with or without UCB and LP. TER (Ω/cm2) = (total resistance − blank resistance) (Ω)/area (cm2).
Immunofluorescence microscopy
Caco-2 cells were grown on glass cover slips, treated as described above, and then rinsed three times with 0·01 m-PBS (pH 7·4). Cells were fixed in 4 % paraformaldehyde for 20 min at room temperature, permeabilised with 0·2 % Triton X-100 for 15 min and then rinsed again in PBS. Cells were incubated at 4°C overnight with primary antibodies against occludin, ZO-1, claudin-1, claudin-4 and JAM-1. After washing, the cover slips were incubated in FITC-conjugated secondary antibody (Jackson Laboratories, Bar Harbor, ME, USA) for 1 h at room temperature and then mounted. To identify the total number of cells, nuclei were stained with Hoechst dye 33 258. Immunolocalisation of tight junction proteins was observed with a confocal fluorescence microscope (LSM 510; Zeiss, Leipzig, Germany).
FITC-phalloidin staining of F-actin was performed as previously described(Reference Dennery, Seidman and Stevenson14). The treated monolayers then were washed with PBS and fixed with 4 % paraformaldehyde in PBS for 30 min. The fixed cells were permeabilised with 0·2 % Triton-X 100 in PBS for 5 min. The cells were washed twice with PBS and then treated with 10 μg/ml of FITC-conjugated phalloidin in PBS for 30 min. After two washes in PBS to remove any traces of non-specific fluorescence, the cells were examined for cytoskeletal actin under a confocal fluorescence microscope.
Detection of protein kinase C and UDP-glucuronosyltransferases 1 family-polypeptide A1 by labelled streptavidin biotin immunocytochemistry
Monolayers of cells were prepared on glass cover slips placed in twelve-well cell culture clusters (Corning Inc.). After washing in PBS, cells were fixed for 15 min at room temperature in 4 % paraformaldehyde and then washed. Endogenous peroxidase was blocked with H2O2 (30 ml/l) for 15 min; non-specific binding sites were blocked with 5 % normal goat serum. Cell monolayers were incubated with a specific primary antibody overnight at 4°C, and primary antibodies were diluted 1:50 (rabbit polyclonal anti-human PKC, rabbit polyclonal anti-human UGT1A1; Santa Cruz Biotechnology) in PBS. Monolayers then were washed three times with PBS for 5 min each time, followed by incubation with biotinylated goat anti-rabbit IgG at 37°C for 30 in. After being washed with PBS as before, HRP-labelled streptavidin was added. The following incubation and washing were exactly the same as above. Finally, DAKO working solution was added for colour development. The reaction was stopped with a tap water rinse. The sections then were counterstained with haematoxylin and mounted.
Western blot analysis
Caco-2 cells were grown to 95 % confluence, treated as described above and then rinsed three times with chilled 0·01 m-PBS (pH 7·4). Proteins were then extracted with 200 μl of ice-cold radioimmunoprecipitation assay (RIPA) buffer (150 mm-NaCl, 50 mm-2-amino-2-hydroxymethyl-propane-1,3-diol (Tris)-HCl (pH 7·4), 0·5 mm-phenylmethylsulfonyl fluoride, 2·4 mm-EDTA and 1 mm-sodium orthovanadate, with 1 % Nonidet-40 (NP-40) and Sigma protease inhibitor cocktail (1:100)) for 30 min at 4°C. After centrifugation at 10 000 g for 10 min at 4°C, the protein concentration of each sample was quantified using the Bradford method. Equal amounts of total protein were separated on 10 % SDS-polyacrylamide gels and then transferred to a nitrocellulose membrane. After blocking overnight in Tris-buffered saline (TBS) containing 0·05 % Tween (TBS-T) and 5 % dry powdered milk, membranes were washed three times for 5 min each with TBS-T and incubated for 2 h at room temperature with primary antibodies against occludin, ZO-1, claudin-1, claudin-4, JAM-1, PKC and UGT1A1. After three washes with TBS-T, the membranes were incubated for 1 h with HRP-conjugated secondary antibody. Following two washes with TBS-T and one wash with TBS, the membranes were developed for visualisation of protein by the addition of an enhanced chemiluminescence reagent (Amersham, Princeton, NJ, USA). Densitometric analysis was performed using the Alpha Imager 1220 system (Alphainotech Co., San Leandro, CA, USA).
Real-time fluorescent quantitative PCR assay
The levels of occludin, ZO-1, claudin-1, claudin-4, JAM-1, PKC and UGT1A1 mRNA were measured using the real-time RT-PCR method with SYBR1 green dye. Total RNA was isolated from Caco-2 cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. Real-time PCR was performed with an ABI prism 7000 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Primers were designed using the Primer Express® program (Applied Biosystems); the sequences are shown in Table 1. The following procedure used 2 μg RNA. In a sterile RNase-free microcentrifuge tube, 1 μl of 20 μm oligo (dT)15 primer was added to a total volume of 15 μl in water. The tube was heated to 70°C for 5 min to melt secondary structures within the template. Next, the tube was cooled immediately on ice to prevent secondary structures from reforming, and then it was spun briefly to collect the solution at the bottom of the tube. The following components were added to the annealed primer/template: 5 μl of 5 × Maloney murine leukaemia virus (M-MLV) reaction buffer, 1·25 μl of 10 mm-dNTPs and twenty-five units of RNasin RNAse inhibitor. Further, 200 units of M-MLV RT RNAse H- were added to the reagent to obtain a 25 μl total reaction volume. The mixture was mixed gently by flicking the tube and then it was incubated for 60 min at 42°C before termination of the reaction at − 20°C.
Table 1 Sequences of oligonucleotide primers and conditions for real-time PCR

ZO, zonula occludens; JAM, junctional adhesion molecule; PKC, protein kinase C; UGT1A1, UDP-glucuronosyltransferases 1 family-polypeptide A1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
* Primers were designed based on sequences of human corresponding genes from the GenBank database.
Glyceraldehyde-3-phosphate dehydrogenase served as the housekeeping gene control. Separate PCR reactions (25 μl) were conducted for each transcript and consisted of cDNA (2·0 μl), 12·5 μl of 2 × SYBR Premix Ex Taq™ (TaKaRa, Ltd, Shiga, Japan) and 0·5 μl each of 10 μm gene-specific forward and reverse primers. PCR conditions were optimised to 95°C (over 30 s), followed by forty cycles (45 s each) of 10 s at 95°C, 5 s at 60°C and 30 s at 72°C. The reaction was completed with 30 s at 37°C. Five serial dilutions of cDNA were analysed for each target gene and were used to construct linear standard curves. To compensate for variations in RNA input and in the efficiency of quantitative RT-PCR, we used a normalisation strategy based on glyceraldehyde-3-phosphate dehydrogenase. The raw data for the expression of each target gene were divided by the quantity of glyceraldehyde-3-phosphate dehydrogenase present to obtain the normalised value of the yield expressed in arbitrary units.
Immunoprecipitation and immunoblotting assays
Caco-2 cells were grown to 95 % confluence, given the appropriate treatment for 6 h and then rinsed three times with chilled 0·01 m-PBS (pH 7·4). Protein then was extracted with 200 μl of ice-cold RIPA buffer (150 mm-NaCl, 50 mm-Tris-HCl (pH 7·4), 0·5 mm-phenylmethylsulfonyl fluoride, 2·4 mm-EDTA and 1 mm-sodium orthovanadate, with 1 % Nonidet-40 (NP-40) and Sigma protease inhibitor cocktail (1:100)) for 30 min at 4°C. After centrifugation at 10 000 g for 10 min at 4°C, the protein concentration of each sample was quantified using the Bradford method. The supernatant fraction was pre-cleared with protein G plus protein A agarose beads (Sigma, St Louis, MO, USA) and incubated overnight at 4°C with rabbit anti-occludin antibody (Zymed) and protein G+ protein A agarose beads. The beads were washed with PBS and ice-cold RIPA buffer. The immunoprecipitated proteins were separated on a 10 % SDS-PAGE gel and transferred onto a nitrocellulose membrane (Invitrogen). The membranes were blocked with 1 % bovine serum albumin in PBS overnight at 4°C and incubated with rabbit anti-phosphoserine antibodies (Zymed) for 2 h at room temperature, then treated with the HRP-conjugated secondary antibody (Santa Cruz Biotechnology). The reaction was visualised using an ECL kit from Pierce (Rockford, IL, USA), and Western blotting was performed with the anti-occludin rabbit polyclonal antibody (Zymed) followed by the anti-rabbit secondary antibody coupled with peroxidase (Santa Cruz Biotechnology) and ECL. For Western blotting of ZO-1, the same protocol was used with the rabbit polyclonal anti-ZO-1 antibody (Santa Cruz Biotechnology) and the rabbit anti-β-actin antibody (Santa Cruz Biotechnology).
Protein kinase C activity assay
The PKC activity assay was conducted following the instructions of the PepTag non-radioactive PKC assay kit (Promega, Madison, WI, USA). Briefly, the cells were washed once with PBS and then lysed in a lysis buffer that included 20 mm-Tris-HCl, 0·5 mm-EGTA, 2 mm-EDTA, 2 mm-phenylmethylsulfonyl fluoride and leupeptin (10 mg/l) (pH 7·5). Assays were then performed at 30°C in a total volume of 25 μl containing 5 μl PKC reaction 5 × buffer, 5 μl PLSRTLSVAAK peptide, 5 μl PKC activator, 1 μl peptide protection solution and 9 μl sample. Reactions were initiated by the addition of the 9 μl sample and terminated after 30 min by incubation of the reaction mixture at 95°C for 10 min. After adding 1 μl of 80 % glycerol, each sample was separated via 0·8 % agarose gel electrophoresis at 100 V for 15 min. The intensity of the fluorescence of the phosphorylated peptides reflected the activity of PKC. All experiments were carried out in triplicate, with each data point representing a separate culture. The experiments yielded similar results each time.
Statistical analysis
Results are presented as the mean values and standard deviations of a minimum of three experiments for each result. The data were analysed using GraphPad PRISM (GraphPad Software Inc., San Diego, CA, USA) and the SPSS 11.0 statistical software package (SPSS Inc., Chicago, IL, USA). All data were analysed using one-way ANOVA with Bonferroni–Dunnett's T3 post hoc multiple comparisons to test for differences between each pair of experimental groups. The difference was considered significant when at P < 0·05.
Results
Viability of Caco-2 cells exposed to unconjugated bilirubin
Caco-2 cell monolayers were exposed to increasing concentrations of UCB for 6 h, and cytotoxicity was investigated using the WST-8 colorimetric assay. Growth of cells was significantly inhibited by UCB in a concentration-dependent manner as the UCB concentration increased from 200 nm to 1000 nm. However, there were no significant differences in Caco-2 cell growth inhibition in the UCB concentration range of 10–100 nm (Fig. 1).

Fig. 1 Caco-2 cell monolayers were exposed for 6 h to increasing concentrations of unconjugated bilirubin (UCB), and cytotoxicity was investigated via2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (WST-8) colorimetric analysis. Values are means, with standard deviations represented by vertical bars. Bilirubin did not affect cell viability in the 10–100 nm range († P>0·05). However, significant cytotoxicity was induced by UCB in a concentration-dependent manner at UCB concentrations from 200 to 1000 nm (* P < 0·05).
Assessment of Caco-2 apoptosis using fuzzy c-means
Annexin V–FITC analysis by fuzzy c-means (FCM) was used to determine the percentage of apoptotic cells after exposure to UCB (1000 nm) with or without LP (108 CFU/ml). The sum of the upper right (UR) and lower right (LR) quadrants represents the number of apoptotic cells in the early stage. The percentage of apoptotic cells increased significantly after treatment for 6 h with 1000 nm-UCB. In the groups co-incubated with LP, the percentage of apoptotic cells decreased significantly as compared with the UCB group. There were no significant differences between the LP before UCB group (group B) and the LP following UCB group (group C) (Fig. 2).

Fig. 2 Annexin V–fluorescein isothiocyanate (FITC) analysis by fuzzy c-means (FCM) showed the percentage of apoptotic cells after exposure to unconjugated bilirubin (UCB) (1000 nm) with or without Lactobacillus plantarum (LP) (108 colony-forming units/ml). (![]() ), UCB only, group A; (
), UCB only, group A; (![]() ), LP before UCB, group B; (□), LP following UCB, group C; (
), LP before UCB, group B; (□), LP following UCB, group C; (![]() ), control, group D. Values are means, with standard deviations represented by vertical bars. Treatment with UCB led to a high percentage of apoptotic cells. However, co-incubation with LP partly inhibited apoptosis. There were no significant differences based on the addition of LP before UCB and following UCB. * Mean value was significantly different from those of the other three groups (group D v. group A, group B, group C) (P < 0·05). † Mean value was not significantly different from that of the LP following UCB group (P>0·05).
), control, group D. Values are means, with standard deviations represented by vertical bars. Treatment with UCB led to a high percentage of apoptotic cells. However, co-incubation with LP partly inhibited apoptosis. There were no significant differences based on the addition of LP before UCB and following UCB. * Mean value was significantly different from those of the other three groups (group D v. group A, group B, group C) (P < 0·05). † Mean value was not significantly different from that of the LP following UCB group (P>0·05).
Lactobacillus plantarum attenuates the unconjugated bilirubin-induced decrease in transepithelial electrical resistance of Caco-2 cells
Caco-2 cells were incubated for 6 h with or without UCB (50 nm) and LP (108 CFU/ml). TER levels decreased significantly after treatment for 6 h with 50 nm-UCB. Co-incubation with LP significantly reduced the toxicity of UCB and restored the TER of the monolayer to some extent. Again, there were no significant differences between the LP before UCB group and the LP following UCB group (Fig. 3).

Fig. 3 Lactobacillus plantarum (LP) attenuates unconjugated bilirubin (UCB)-induced decreases in the transepithelial electrical resistance (TER) of Caco-2 cells. (![]() ), UCB only; (
), UCB only; (![]() ), LP before UCB; (
), LP before UCB; (![]() ), LP following UCB; (
), LP following UCB; (![]() ), control. Values are means, with standard deviations represented by vertical bars. TER was significantly lower after UCB treatment compared with the control group. Co-incubation with LP partly restored the TER of the monolayers. There were no significant differences in results based on the addition of LP before or following UCB administration. * Mean value was significantly different from those of the other three groups (P < 0·05).
), control. Values are means, with standard deviations represented by vertical bars. TER was significantly lower after UCB treatment compared with the control group. Co-incubation with LP partly restored the TER of the monolayers. There were no significant differences in results based on the addition of LP before or following UCB administration. * Mean value was significantly different from those of the other three groups (P < 0·05).
Detection of protein kinase C and UDP-glucuronosyltransferases 1 family-polypeptide A1 expression by immunocytochemistry
PKC and UGT1A1 were observed as brown spots in the perinuclear structure. In the Caco-2 cells incubated with UCB, expression of UGT1A1 was higher than that of the control. In the group co-incubated with LP, the number of brown spots was lower compared with the UCB group; however, expression was more prominent than in the normal control group. There were no significant differences between groups B and C. Conversely, the expression of PKC decreased when Caco-2 cells were treated with UCB; however, the addition of LP increased PKC expression. No differences were observed between groups B and C (Fig. 4).

Fig. 4 Immunohistochemical expression of protein kinase C (PKC) and UDP-glucuronosyltransferases 1 family-polypeptide A1 (UGT1A1) in Caco-2 cells. In the control group, immunoreactivity for PKC was stronger than it was in the unconjugated bilirubin (UCB) group. In both Lactobacillus plantarum (LP) addition groups, PKC expression was nearly restored to the control level. UCB induced significant expression of UGT1A1 in the UCB-only group, whereas no staining was observed in the control group. Administration of LP before UCB and following UCB made no difference in the UGT1A1 expression.
Effects of Lactobacillus plantarum and unconjugated bilirubin on tight junction protein localisation
Confocal imaging was performed to assess the distribution of tight junctions after exposure to UCB (50 nm) and LP (108 CFU/ml). Tight junction-associated proteins were continuously distributed with bright green or red spots along membranes of the control cells. Occludin, claudin-1, claudin-4 and JAM-1 were located at the outer edge of the membrane, whereas ZO-1 was distributed in the cytoplasm; the borders of the cells were very clear in the control group. In the control Caco-2 intestinal monolayers, tight junction-associated proteins were present at the apical intercellular borders in a belt-like manner, encircling the cells and delineating the cellular borders. In group A (the USB-treated group), the fluorescence was dispersed and even became punctate, with some loss from the membrane (in contrast to the uniform membrane staining in controls). In the groups co-incubated with LP, the amount of tight junction-associated proteins was decreased compared with that of the control group; however, protein expression was increased relative to group A. There were no significant differences between groups B and C with respect to proteins.
The F-actin staining pattern of control Caco-2 cells showed a continuously lined distribution at the cell borders and cytoskeletal regions. A high density of actin filaments was present at the apical perijunctional regions, encircling the cells in a belt-like manner. In contrast, the actin architecture in group A was disorganised and disrupted. The UCB-induced alteration of F-actin filaments was reversed by co-incubation with LP. No differences were observed when comparing groups B and C (Fig. 5(a) and (b)).

Fig. 5 (a) Immunofluorescent detection of zonula occludens (ZO)-1, claudin-4 and junctional adhesion molecule (JAM)-A in Caco-2 cells. The staining intensity of the unconjugated bilirubin (UCB)-treated cells was decreased compared with that of control cells. Lactobacillus plantarum (LP) prevented UCB-induced redistribution of the above-mentioned tight junction proteins. (b) Immunofluorescent detection of occludin, F-actin and claudin-1 in Caco-2 cells. The staining intensity of the UCB-treated cells was decreased compared with that of control cells. LP prevented UCB-induced redistribution of the above-mentioned tight junction proteins.
Effects of Lactobacillus plantarum and unconjugated bilirubin on tight junction proteins, protein kinase C and UDP-glucuronosyltransferases 1 family-polypeptide A1 protein expression (Western blot)
Western blot analyses were performed to determine the protein expression levels of occludin, claudin-1, claudin-4, JAM-1, ZO-1, PKC and UGT1A1 in Caco-2 cells after treatment with UCB and with or without LP. The intensity measurements for whole-cell proteins were determined from the ratio of the integrated band intensity of the target proteins to that of β-actin in the same sample. Western blotting of epithelial whole-cell protein extracts showed that the expression of target proteins was reduced in UCB-treated cells as compared with the control group (group A v. group D). There was an increase in the tight junction protein expression density in the LP group as compared with the UCB group (P < 0·05). As was expected, UCB induced the expression of UGT1A1 proteins in Caco-2 cells; however, UGT1A1 protein was not affected by LP (Fig. 6(a) and (b)).

Fig. 6 (a) Effects of unconjugated bilirubin (UCB) and Lactobacillus plantarum (LP) on the expression of tight junction proteins and protein kinase C (PKC) and UDP-glucuronosyltransferases 1 family-polypeptide A1 (UGT1A1) proteins in Caco-2 cell monolayers. Western blotting analysis of occludin, zonula occludens (ZO)-1, claudin-1, claudin-4, junctional adhesion molecule (JAM)-1, PKC and UGT1A1 proteins. (b) Statistical evaluation of densitometric data representing protein expression in three separate experiments. (![]() ), UCB only; (
), UCB only; (![]() ), LP before UCB; (□), LP following UCB; (
), LP before UCB; (□), LP following UCB; (![]() ), control. Values are means, with standard deviations represented by vertical bars. UCB triggered a decrease in the tight junction proteins, and PKC simultaneously induced the expression of UGT1A1 proteins. The addition of LP increased expression of tight junction proteins and PKC, but it did not affect UGT1A1 protein expression. * Mean value was significantly different from those of the other three groups (P < 0·05). † Mean value was not significantly different from that of the LP following UCB group (P>0·05).
), control. Values are means, with standard deviations represented by vertical bars. UCB triggered a decrease in the tight junction proteins, and PKC simultaneously induced the expression of UGT1A1 proteins. The addition of LP increased expression of tight junction proteins and PKC, but it did not affect UGT1A1 protein expression. * Mean value was significantly different from those of the other three groups (P < 0·05). † Mean value was not significantly different from that of the LP following UCB group (P>0·05).
Expression of mRNA for tight junction proteins, protein kinase C and UDP-glucuronosyltransferases 1 family-polypeptide A1 as detected by real-time fluorescent quantitative PCR
Treatment of Caco-2 cells with LP and UCB resulted in altered levels of occludin, ZO-1, claudin-1, claudin-4 and JAM-1, PKC and UGT1A1. This result caused us to speculate that these altered protein levels might result from changes in mRNA level. We therefore used quantitative RT-PCR to determine the level of mRNA in each group of Caco-2 cells. The levels of mRNA of occludin, ZO-1, claudin-1, claudin-4, JAM-1 and PKC decreased significantly after exposure to UCB (50 nm). Upon co-incubation with LP, the levels of mRNA of the target proteins increased. There were no significant differences between groups B and C; however, the level of UGT1A1 mRNA increased significantly after exposure to UCB (50 nm) (group A v. group D). In the groups co-incubated with LP, the levels of mRNA coding UGT1A1 proteins were increased, and there were no significant differences between these groups (B and C) (Table 2, Fig. 7).
Table 2 Expression (mRNA) ratio (studied genes:glyceraldehyde-3-phosphate dehydrogenase (GAPDH)) for tight junction proteins and UDP-glucuronosyltransferases 1 family-polypeptide A1 (UGT1A1) in Caco-2 cells induced by unconjugated bilirubin (UCB) co-cultured with or without Lactobacillus plantarum (LP) for 6 h
(Mean values and standard deviations)
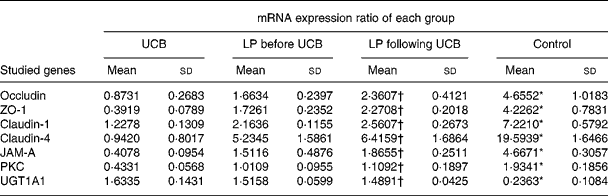
ZO, zonula occludens; JAM, junctional adhesion molecule; PKC, protein kinase C.
* Mean value was significantly different from those of the other three groups (P < 0·05).
† Mean value was not significantly different from that of the LP before UCB group (P>0·05).

Fig. 7 SYBR green-based real-time quantitative RT-PCR techniques were used to measure mRNA expression ratios (studied genes:glyceraldehyde-3-phosphate dehydrogenase (GAPDH) ratio) for tight junction proteins, protein kinase C (PKC) and UDP-glucuronosyltransferases 1 family-polypeptide A1 (UGT1A1) in Caco-2 cells of all four groups. (![]() ), Unconjugated bilirubin (UCB) only; (
), Unconjugated bilirubin (UCB) only; (![]() ), Lactobacillus plantarum (LP) before UCB; (□), LP following UCB; (
), Lactobacillus plantarum (LP) before UCB; (□), LP following UCB; (![]() ), control; ZO, zonula occludens; JAM, junctional adhesion molecule. Values are means, with standard deviations represented by vertical bars. * Mean value was significantly different from those of the other three groups (P < 0·05). † Mean value was not significantly different from that of the LP following UCB group (P>0·05).
), control; ZO, zonula occludens; JAM, junctional adhesion molecule. Values are means, with standard deviations represented by vertical bars. * Mean value was significantly different from those of the other three groups (P < 0·05). † Mean value was not significantly different from that of the LP following UCB group (P>0·05).
Phosphorylation of occludin and zonula occludens-1
We examined the phosphorylation status of occludin and ZO-1 by immunoprecipitation and immunoblotting assays. Occludin and ZO-1 were found to be phosphorylated at their serine residues. Incubation with UCB resulted in decreased phosphorylated occludin and phosphorylated ZO-1 protein expression when compared with untreated cells, whereas the addition of LP increased the expression of phosphorylated occludin and phosphorylated ZO-1. Again, no significant differences were detected between groups B and C (Fig. 8(a) and (b)).

Fig. 8 (a) Serine phosphorylation of occludin (p-occludin) and zonula occludens (ZO)-1 (p-ZO-1) in Caco-2 cells. Cell lysates were subjected to immunoprecipitation with the anti-occludin or ZO-1 antibody, followed by Western blot analysis with antibodies against phosphorylated serine. UCB, unconjugated bilirubin; LP, Lactobacillus plantarum. (b) Statistical evaluation of serine phosphorylation of occludin and ZO-1 in Caco-2 cells. Average results in a group of cells showing the UCB-induced decreases in p-occludin and p-ZO-1 protein expression as compared with untreated cells. (![]() ), UCB only; (
), UCB only; (![]() ), LP before UCB; (□), LP following UCB; (
), LP before UCB; (□), LP following UCB; (![]() ), control. Values are means, with standard deviations represented by vertical bars. While the addition of LP increased the expression of the p-occludin and p-ZO-1 proteins, there were no significant differences between LP administered before or after UCB administration. * Mean value was significantly different from those of the other three groups (P < 0·05). † Mean value was not significantly different from that of the LP following UCB group (P>0·05).
), control. Values are means, with standard deviations represented by vertical bars. While the addition of LP increased the expression of the p-occludin and p-ZO-1 proteins, there were no significant differences between LP administered before or after UCB administration. * Mean value was significantly different from those of the other three groups (P < 0·05). † Mean value was not significantly different from that of the LP following UCB group (P>0·05).
Effects of Lactobacillus plantarum and unconjugated bilirubin on the activity of protein kinase C
As shown in Fig. 9(a) and (b), the addition of UCB (50 mm) to the culture medium resulted in a significant decrease in PKC activity. Co-incubation with LP partly restored PKC activity in both groups B and C, but there were no significant differences between these two groups.

Fig. 9 (a) Effects of unconjugated bilirubin (UCB) and Lactobacillus plantarum (LP) on the activity of protein kinase C (PKC) in Caco-2 cells. Representative gel electrophoresis from the PKC activity assay. (b) Statistical evaluation of UCB and LP on the activity of PKC by average results in a group of Caco-2 cells. (![]() ), UCB only; (
), UCB only; (![]() ), LP before UCB; (□), LP following UCB; (
), LP before UCB; (□), LP following UCB; (![]() ), control. Values are means, with standard deviations represented by vertical bars. * Mean value was significantly different from those of the other three groups (P < 0·05). † Mean value was not significantly different from that of the LP following UCB group (P>0·05).
), control. Values are means, with standard deviations represented by vertical bars. * Mean value was significantly different from those of the other three groups (P < 0·05). † Mean value was not significantly different from that of the LP following UCB group (P>0·05).
Discussion
Numerous research groups have suggested that hyperbilirubinaemia is associated with a disruption of intestinal integrity. However, few studies have examined the relationship between bilirubin and the intestinal mucosal barrier. Conjugated bilirubin and UCB, when bound to serum albumin, cannot cross membranes or interfere with cells. However, the enzyme glycuronidase, of both endogenous and microbial origin, partially converts UCB to conjugated bilirubin(Reference Dennery, Seidman and Stevenson14). Because UCB can cause severe damage to the central nervous system, most investigators have focused their research on its effects on neural cells. At the intracellular level, UCB interferes with DNA and protein synthesis(Reference Yamada, Sawasaki and Nakajima15), inhibits protein kinases and protein phosphorylation(Reference Hansen, Mathiesen and Walaas16), and causes lactate dehydrogenase (LDH) release and apoptosis in neurons(Reference Rodrigues, Solá and Brites17). Raimondi et al. recently investigated the effects of UCB on the barrier function of human intestinal Caco-2 cell monolayers and reported that bilirubin induced a concentration-dependent decrease of TER(Reference Raimondi, Crivaro and Capasso2). Furthermore, bilirubin at a 50 nmol/l concentration triggered a reversible redistribution of tight junctional occluding, and a UCB concentration of 600 nmol/l was significantly cytotoxic.
Many clinical studies have reported that probiotics such as LP have beneficial health effects(Reference Huebner and Surawicz18). A recent study reported that LP protected Caco-2 monolayer integrity and the distribution of tight junction proteins(Reference Qin, Zhang and Hang19). Surface-layer protein extracts from L. helveticus inhibited enterohaemorrhagic Escherichia coli adhesion to epithelial cells and protected the intestinal epithelial functions of monolayers(Reference Johnson-Henry, Hagen and Gordonpour20). Enteral administration of the probiotic bacterium LP299 reduced intestinal hyperpermeability associated with experimental biliary obstruction(Reference White, Hoper and Parks21).
The present study demonstrated that growth of Caco-2 cells was significantly inhibited by cytotoxic doses of UCB in a concentration-dependent manner. This finding is in agreement with a previous study published by Raimondi et al. (Reference Raimondi, Crivaro and Capasso2). The present study also demonstrated that UCB disrupted epithelial tight junction structure, including the distribution of proteins such as occludin, ZO-1, claudin-1, claudin-4, JAM-1 and F-actin, in cultured Caco-2 cells, resulting in decreased TER. In the present study, LP mitigated the UCB-induced redistribution of occludin, ZO-1, claudin-1, claudin-4, JAM-1 and F-actin. We also demonstrated that LP treatment stabilised cellular tight junctions and lessened UCB cytotoxicity, thereby preventing UCB-induced redistribution of the integral tight junction proteins and the apoptosis of Caco-2 cells. To support the microscopy observations, we also used Western blotting techniques and quantitative RT-PCR techniques to measure levels of occludin, ZO-1, claudin-1, claudin-4 and JAM-1. Co-incubation with LP resulted in partial restoration of tight junction proteins and of the mRNA of these proteins and helped to maintain the morphology of Caco-2 cells.
Little is known about the mechanisms underlying UCB-induced tight junction disruption in the Caco-2 cell monolayer or the mechanism responsible for the reversible toxicity of UCB. A previous study suggested that UCB oxidation might explain the reversible nature of the permeability increase. As a complementary hypothesis, some of the UCB molecules might be pumped out of the cell by carriers known to be expressed in this cell line(Reference Tiribelli and Ostrow22). UGT1A1-dependent bilirubin conjugation plays a critical role in the detoxification of UCB. To explore the possibility of induction of UGT1A1 expression by UCB in Caco-2 cells, we used immunocytochemistry, Western blotting and quantitative RT-PCR techniques to determine the expression levels of UGT1A1. The present study supported the hypothesis that UCB itself can induce UGT1A1 expression in Caco-2 cells and then decrease UCB concentration to relatively low levels, finally reducing the cytotoxicity of UCB. LP treatment did not change the expression of UGT1A1 induced by UCB in the present study.
Hansen et al. reported that the widespread inhibitory effect of bilirubin on protein kinases may contribute to bilirubin neurotoxicity(Reference Hansen, Mathiesen and Walaas16). A substantial body of experimental data indicates that PKC regulates paracellular permeability in epithelial cells(Reference Rosson, O'Brien and Kampherstein23, Reference Mullin, Kampherstein and Laughlin24), and PKC also regulates the assembly of tight junction proteins through phosphorylation of ZO-1(Reference Stuart and Nigam25). It also has been demonstrated that (i) inhibition of PP2A by okadaic acid promotes the phosphorylation and recruitment of ZO-1, occludin and claudin-1 to the tight junction in MDCK cells in an a PKC-dependent manner(Reference Nunbhakdi-Craig, Machleidt and Ogris26), (ii) highly phosphorylated occludin is selectively concentrated at tight junctions, whereas non-phosphorylated or less phosphorylated occludin is distributed in the basolateral membrane and in the cytoplasm(Reference Sakakibara, Furuse and Saitou27), and (iii) PKC phosphorylates occludin at threonine residues (T403 and T404) and plays a crucial role in the assembly and/or maintenance of tight junctions in Caco-2 and MDCK cell monolayers(Reference Suzuki, Elias and Seth28). Probiotic-secretory proteins protect the intestinal epithelial tight junctions and barrier from H2O2-induced insult by a PKC- and MAP kinase-dependent mechanism(Reference Seth, Yan and Polk29).
The present study confirmed the disruption of the intestinal barrier by UCB and that administration of LP is associated with restored intestinal integrity after disruption by UCB. A key question that remains is whether the observed protective effects are due to UCB detoxification by the microbe. Although Goldin & Gorbach investigated the effect of oral supplements of L. acidophilus on human faecal bacterial enzyme activity and reported that the activity of β-glucuronidase was 2- to 4-fold reduced in the L. acidophilus-treated compared with controls(Reference Goldin and Gorbach30), no report currently exist about probiotics themselves taking part directly in the metabolism of bilirubin. To clarify whether PKC mediates the disruption of the intestinal barrier induced by UCB, we examined the phosphorylation status of occludin and ZO-1 using a Western blot analysis with antibodies against phosphorylated serine. We found that UCB treatment decreased phosphorylated occludin and phosphorylated ZO-1 protein expression compared with untreated cells, while the addition of LP increased the expression of phosphorylated occludin and phosphorylated ZO-1. There were no significant differences between the group that received LP before UCB and the group that received LP following UCB. We found that UCB in the culture medium resulted in a significant decrease in PKC activity. Co-incubation with LP partly restored PKC activity in both LP groups (LP first and UCB first).
In conclusion, the present short study supports the hypothesis that the administration of LP is associated with restored intestinal integrity after disruption by UCB. Furthermore, UGT1A1 may be involved in the reversibility of changes in the intestinal mucosal barrier after 24–48 h of treatment with 50 nm-UCB. The present study also demonstrated that UCB inhibits PKC activity and decreases phosphorylated occludin and phosphorylated ZO-1 protein expression. Our findings suggest that LP protects UCB-induced tight junction disruption by activating the PKC pathway; however, further research is warranted to confirm these conclusions.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (no. 30471687) and the Ministry of Science and Technology of the People's Republic of China (no. 2008CB517403).
The authors' contributions were as follows: Y. Z. carried out the study, participated in its design, was responsible for data collection and sample analyses, and wrote the original manuscript. H. Q. participated in the study design, helped draft the manuscript and acquired the funding. M. Z. conducted the gel electrophoresis and Western blotting. T. S. participated in the study design. Y. M., H. C. and Z. C. participated in the immunohistochemistry and fluorescence staining. P. Z. and Z. L. conducted document retrieval and data analysis. All authors read and approved the findings of the study.
The authors declare that there are no personal or financial conflicts of interest associated with this paper.











