


Lipoprotein(a) (Lp(a)) is an LDL-like particle bound to apolipoprotein (a)(Reference Ellis, Boffa and Sahebkar1). Increasing evidence supports elevated Lp(a), which may be defined as >75 nmol/l or 30 mg/dl, for being an independent and important risk factor for CVD(Reference Nordestgaard, Chapman and Ray2). The plasma level of Lp(a) is considered to be determined mostly by the LPA gene locus(Reference Kronenberg and Utermann3); however, lifestyle-induced changes in the plasma level of Lp(a) have been reported(Reference Berk, Yahya and Verhoeven4–Reference Silaste, Rantala and Alfthan8).
Dietary fatty acids (FA) differently impact CVD risk through divergent effects on the lipid profile(Reference Wang and Hu9). Despite a large body of evidence documenting the beneficial effect of replacing SFA with PUFA in the prevention of CVD(Reference Mozaffarian, Micha and Wallace10,Reference Hooper, Martin and Abdelhamid11) , recent data from Chowdhury et al. and Ramsden et al. have reported conflicting results and concluded that there is no clear support for replacing SFA with PUFA(Reference Chowdhury, Warnakula and Kunutsor12,Reference Ramsden, Zamora and Majchrzak-Hong13) . However, in 2017, the American Heart Association presidential advisory on dietary fats and CVD strongly concluded that the incidence of CVD will be reduced when the intake of SFA is replaced with PUFA(Reference Sacks, Lichtenstein and Wu14). Nevertheless, one of the cornerstones in diet recommendations when performing lifestyle changes and in the prevention of CVD is replacing SFA with PUFA(Reference Piepoli, Hoes and Agewall15). The essential n-6 PUFA linoleic acid is the predominant dietary PUFA, and lower tissue/blood concentrations of linoleic acid have been shown to be inversely associated with CVD risk(Reference Harris, Poston and Haddock16) and mortality(Reference Virtanen, Wu and Voutilainen17). Furthermore, Marklund et al. recently concluded that higher circulating/tissue levels of linoleic acid and possibly arachidonic acid (AA) were inversely associated with the risk of major cardiovascular events after analysing thirty prospective studies from thirteen countries(Reference Marklund, Wu and Imamura18). However, some inconsistent data have also been reported. In the reanalysis of the Sydney Diet Heart study, it was shown that the intervention group had higher rates of death than controls(Reference Ramsden, Zamora and Leelarthaepin19); and in a Mendelian randomisation study, genetically predicted linoleic acid was shown not to be associated with ischemic heart disease but was associated with lower diabetes risk(Reference Zhao and Schooling20). Recently, Berk et al. found increased Lp(a) levels elicited by diet-induced weight loss after a low-fat diet in overweight and obese subjects(Reference Berk, Yahya and Verhoeven4). However, they found no change in Lp(a) levels after bariatric surgery(Reference Berk, Yahya and Verhoeven4), suggesting a role of dietary FA in the regulation of Lp(a), rather than weight loss per se. Indeed, randomised controlled trials (RCT) have found Lp(a)-increasing effects of low-fat v. low carbohydrate diets(Reference Faghihnia, Tsimikas and Miller5–Reference Silaste, Rantala and Alfthan8).
Subjects with familial hypercholesterolaemia (FH) are characterised by increased plasma levels of total and LDL-cholesterol, accelerated atherosclerosis and increased risk of premature CVD(Reference Nordestgaard, Chapman and Humphries21,Reference Mundal, Veierod and Halvorsen22) and may have higher Lp(a) levels than unaffected relatives(Reference Alonso, Andres and Mata23). Thus, FH subjects may serve as a suitable human model to investigate the underlying mechanisms of Lp(a) modification.
To our knowledge, no studies have investigated whether there is an association between Lp(a) levels and n-6 PUFA. Although the magnitude of the effect may not be clinically relevant, this association may be mechanistically interesting in understanding more of the role and the regulation of Lp(a). We aimed to investigate whether plasma Lp(a) concentration was associated with dietary n-6 FA intake and plasma levels of AA in subjects with FH.
Methods
Subjects and study design
In this cross-sectional study, we invited FH subjects (>18 years of age) with or without elevated Lp(a) as defined by Lp(a) ≥ or <75 nmol/l, respectively, who were regularly followed up at the outpatient Lipid Clinic, Oslo University Hospital, Norway. Other inclusion criteria were a definite FH diagnosis as defined by a positive DNA test (genetic FH) or a Dutch Lipid Clinic Network score >8(Reference Nordestgaard, Chapman and Humphries21) (clinical FH) and willingness to give a blood sample. All FH subjects continued with their current lipid-lowering therapy during the study. Exclusion criteria were diabetes mellitus type 1 or pregnant or lactating women. The study visit was coordinated simultaneously with their next prescheduled consultation at the Lipid Clinic. Age- and sex-matched (by percentage) healthy controls were recruited among employees and friends of employees at the Department of Nutrition, University of Oslo, Norway and Oslo University Hospital, Norway. Exclusion criteria for the controls were Lp(a) levels ≥75 nmol/l, cardiovascular or metabolic disease, use of lipid-lowering therapy, severe illness such as cancer in the last 5 years or pregnant or lactating women. All participants were recruited in the period September 2016–September 2017. For all participants, a non-fasting blood sample was obtained, and weight, height and blood pressure were measured. Informed consent was obtained from all the participants. The study protocol was approved by the Regional Committee of Medical and Health Research Ethics, south-east region of Norway (no. 2015/1577) and by the Privacy Ombudsman at Oslo University Hospital. The study was conducted in accordance with the principles of the Declaration of Helsinki.
Total plasma fatty acid profile, fatty acid ratios and other plasma analyses
Plasma was obtained in EDTA tubes and kept in dark on ice until centrifugation at 4°C for 15 min before being aliquoted and stored at –80°C until further analysis. We analysed total plasma FA profile by GC–flame ionisation detector analysis at the commercial laboratory Vitas Analytical Services as previously described(Reference Ulven, Leder and Elind24) and showed the results as percentage of total FA. We estimated certain ratios between the product and the precursor of the individual FA in plasma (γ-linolenic acid (18 : 3n-6)/linoleic acid (18 : 2n-6); AA (20 : 4n-6)/dihomo γ-linolenic acid (20 : 3n-6) and eicosadienoic acid (20 : 2n-6)/linoleic acid (18 : 2n-6)), as described elsewhere(Reference Kawashima, Sugawara and Okita25). The plasma n-6 : n-3 ratio was calculated as the total percentage of n-6 (linoleic acid, γ-linolenic acid, eicosadienoic acid, dihomo γ-linolenic acid and AA) divided by the total percentage of n-3 FA (α-linolenic acid, EPA, n-3 docosapentaenoic acid and DHA). Lp(a) was analysed using an immunoturbidimetric method from Roche Diagnostics at an accredited medical laboratory, Oslo University Hospital, Rikshospitalet, Oslo, Norway (NS-EN ISO 15189 : 2007). At the same medical laboratory, other biochemical analyses were measured in plasma or serum by standard methods.
Dietary intake
Nutrient intake was recorded by a self-administered 256-item food FFQ completed according to a description, both received by mail. In case of incorrect completion, the participants were interviewed at the study visit or through phone. The FFQ is developed at the Department of Nutrition, University of Oslo, Norway, and has been validated and described in detail elsewhere(Reference Carlsen, Lillegaard and Karlsen26). Briefly, intake frequencies (monthly, weekly or daily) and portion sizes in predefined household units were registered. Open spaces were available for description of unlisted food items, which were included in the nutrient calculations. The food database AE-14 and the software ‘Kostberegningssystem’ (version 7.3, 2017) were used to compute the daily intake of energy and nutrients. The food database used (AE-14) is based on the official Norwegian Food Composition Table (http://www.norwegianfoodcomp.no/), supplemented with data from calculated recipes and other databases and has a large number of FA including n-6 PUFA. Total dietary n-6 PUFA is the sum of 18 : 2n-6, 20 : 2n-6, 20 : 3n-6 and 20 : 4n-6.
Statistics
The Kruskal–Wallis test was used for all comparisons between the three groups as regards continuous variables. Pairwise comparisons (post hoc tests) were performed using the Mann–Whitney test and both unadjusted and Bonferroni adjusted corrections were presented. The results are presented as medians and 25th−75th percentiles in the tables and the text and as medians and minimum and maximum values in figures. The χ2 test or Fisher’s exact test was used for categorical data, which are presented as frequencies and percentages in tables. Linear regression analysis was performed to study the association between the dietary intake of total n-6 PUFA, linoleic acid and AA (all in energy percentage (E %)) and plasma AA after adjustment for potential confounding variables among FH patients that completed FFQ. Furthermore, logistic regression analysis was used to adjust for potential confounding variables when studying the association between plasma AA and plasma Lp(a) concentration. Results are presented as regression coefficients and OR, respectively, with 95 % CI. The regression analyses were guided by directed acyclic graphs. Statistical analyses were performed by IBM SPSS Statistics 24.ink. P values (two tailed) <0·05 were considered significant.
Results
Characteristics
In total, FH subjects with (n 68) and without (n 77) elevated Lp(a) levels defined as ≥ and <75 nmol/l plasma Lp(a) and fourteen healthy controls were included in the study (online Supplementary Fig. S1). All the FH subjects had a genetically verified diagnosis except for four who had clinical FH(Reference Nordestgaard, Chapman and Humphries21) (Table 1). All FH patients used lipid-lowering therapy and had generally received dietary counselling as part of their standard follow-up at the lipid clinic. Both groups of FH subjects had significantly higher BMI (P > 0·001) and systolic blood pressure (P = 0·003) than the healthy controls. More FH subjects with elevated Lp(a) levels had experienced CVD (P = 0·02) and were more often treated with proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors (P = 0·01) than the FH subjects without elevated Lp(a). Although non-fasting blood samples were taken, median TAG levels were not elevated according to the European guidelines, where TAG levels ≥1·7 mmol/l is considered elevated(Reference Graham, Atar and Borch-Johnsen27). However, 19·1, 19·5 and 14·3 % of the FH subjects with elevated Lp(a), FH subjects without elevated Lp(a) and controls had TAG levels ≥1·7 mmol/l, respectively.
Table 1. Characteristics of the participants*
(Medians and 25th–75th percentiles; frequencies and percentages)

FH, familial hypercholesterolaemia; Lp(a), lipoprotein(a); SBP, systolic blood pressure; DBP, diastolic blood pressure; Tchol, total cholesterol; HDL-C, HDL-cholesterol; LDL-C, LDL-cholesterol; PCSK9, proprotein convertase subtilisin/kexin type 9.
* P values from the Mann–Whitney U test are shown as unadjusted and Bonferroni adjusted correction.
† Kruskal–Wallis test or χ2 test between the three groups.
‡ Mann–Whitney test or χ2 test between FH subjects Lp(a) ≥ 75 nmol/l and FH subjects Lp(a) < 75 nmol/l.
§ Mann–Whitney test between: FH subjects Lp(a) ≥ 75 nmol/l and controls.
║ Mann–Whitney test between FH subjects Lp(a) < 75 nmol/l and controls.
¶ n 62.
** n 61.
Plasma fatty acid profile and ratios of fatty acids
For plasma levels of n-6 PUFA, we found several significant differences between the groups (Fig. 1). First, we observed higher level of AA (20 : 4n-6) in FH subjects with elevated Lp(a) compared with FH subjects without elevated Lp(a) (P = 0·03; Fig. 1(a)). Furthermore, the levels of AA (20 : 4n-6), dihomo-γ-linolenic acid (20 : 3n-6) and γ-linolenic acid (18 : 3n-6) were all higher in both FH groups compared with the healthy controls (P < 0·001 for all; Fig. 1(a), (b) and (c), respectively). Contrary, the level of linoleic acid (18 : 2n-6) was lower in both FH groups compared with the healthy subjects (P < 0·001 for both; Fig. 1(d)), and the level of eicosadienoic acid (20 : 2n-6) was lower in FH with elevated Lp(a) compared with healthy subjects (P < 0·01; Fig. 1(e)). For the plasma levels of n-3 PUFA (Table 2), we found lower levels of α-linolenic acid (18 : 3n-3) in both FH groups compared with the healthy controls (P < 0·005 for both). The level of DHA (22 : 6n-3) was lower in the FH subjects without elevated Lp(a) than in the healthy controls (P = 0·02). A few significant differences between the groups were also observed among the plasma levels of MUFA and SFA (Table 2).
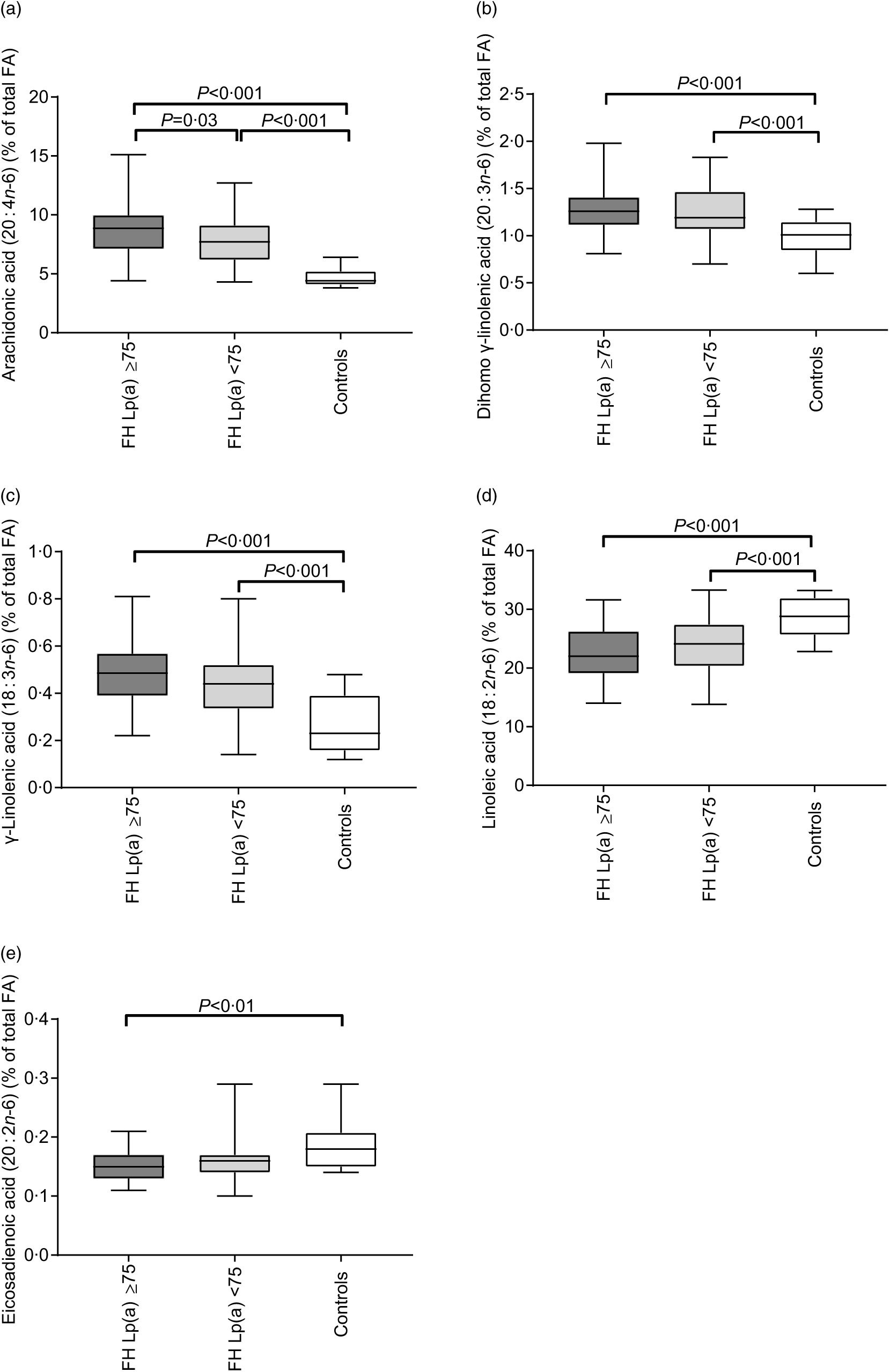
Fig. 1. Plasma levels of arachidonic acid (a), dihomo-γ-linolenic acid (b), γ-linolenic acid (c), linoleic acid (d) and eicosadienoic acid (e) in familial hypercholesterolaemia (FH) subjects with (n 68) or without (n 77) elevated lipoprotein(a) (Lp(a)) and healthy controls (n 14). Data were analysed by the Kruskal–Wallis test with Bonferroni-corrected post hoc comparisons between the groups when significant and given as median (minimum–maximum) percentage of total fatty acids (FA).
Table 2. Fatty acids (FA) in plasma*
(Medians and 25th–75th percentiles)

FH, familial hypercholesterolaemia; Lp(a), lipoprotein(a); ALA, α-linolenic acid; DPA, docosapentaenoic acid.
* P values from the Mann–Whitney U test are shown as unadjusted and Bonferroni adjusted correction.
† Kruskal–Wallis test between the three groups.
‡ Mann–Whitney test between FH subjects Lp(a) ≥ 75 nmol/l and FH subjects Lp(a) < 75 nmol/l.
§ Mann–Whitney test between FH subjects Lp(a) ≥ 75 nmol/l and controls.
║ Mann–Whitney test between FH subjects Lp(a) < 75 nmol/l and controls.
In order to investigate in detail the n-6 PUFA pathway, we estimated the ratios of certain n-6 PUFA (as described in methods). The ratios 18 : 3n-6/18 : 2n-6 and 20 : 4n-6/20 : 3n-6 were higher in both FH groups compared with the healthy controls (P < 0·05 for all; Fig. 2(a)).
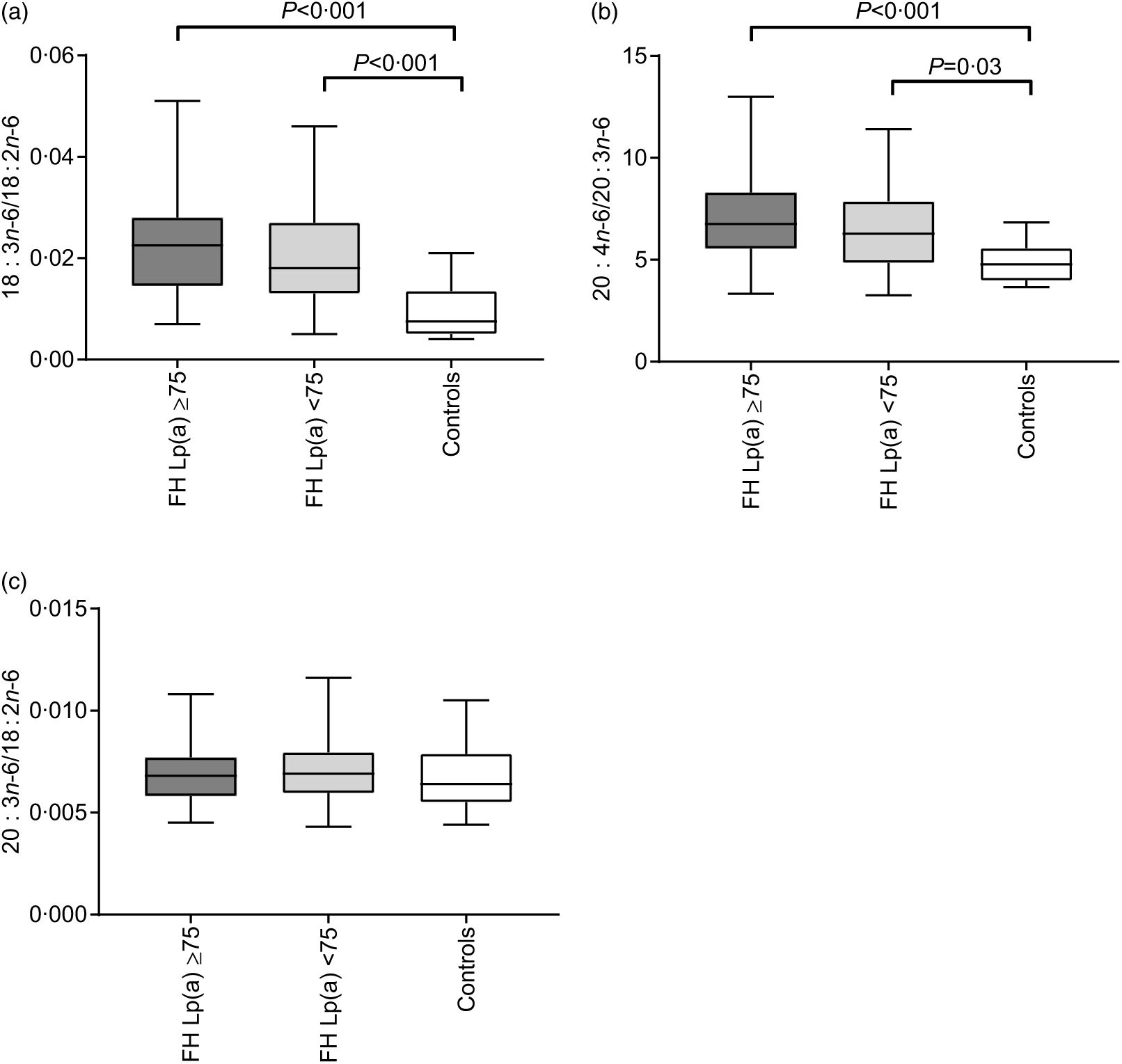
Fig. 2. Estimated ratios as surrogate markers of Δ-6 desaturase (a), Δ-5 desaturase (b) and elongase 5 (c) in familial hypercholesterolaemia (FH) subjects with (n 68) or without (n 77) elevated lipoprotein(a) (Lp(a)) and healthy controls (n 14). The ratios were calculated as product divided by precursor as indicated on the y-axis. Data were analysed by the Kruskal–Wallis test with Bonferroni-corrected post hoc comparisons between the groups when significant and given as median (minimum–maximum).
Dietary intake and dietary pattern
FH subjects with elevated Lp(a) had higher intake of dietary fibre (E %) and lower intake of cholesterol (mg) compared with FH subjects without elevated Lp(a), and lower intake of trans-FA (E %) compared with healthy controls (P = 0·03 for all; Table 3). There was an overall significant difference in SFA intake between the three groups (P = 0·03); however, no significant differences were observed in the pairwise comparisons with Bonferroni adjustment. We found no significant differences between the three groups regarding MUFA, total PUFA, n-3 or n-6 intake (E % for all; Table 3). In order to explain the differences in dietary intake between the FH groups, we explored the dietary patterns of FH subjects with and without elevated Lp(a). Compared with FH subjects without elevated Lp(a), FH subjects with elevated Lp(a) demonstrated lower intake of cakes 7·9 (2·1–18·8) v. 13·6 (7·5–21·4) g/d P = 0·006; sweets 12·0 (4·6–29·1) v. 18·8 (12·0–38·2) g/d P = 0·02, and non-significantly lower intake of cheese 22·2 (12·7–35·1) v. 28·2 (16·0–49·2) g/d P = 0·08. There was no difference in the total intake of food (g/d) between the FH groups (data not shown).
Table 3. Intake of energy and macronutrients*
(Medians and 25th–75th percentiles)

FH, familial hypercholesterolaemia; Lp(a), lipoprotein(a); E %, energy percentage; TFA, trans-unsaturated fatty acids.
* P values from the Mann–Whitney U test are shown as unadjusted and Bonferroni adjusted.
† Kruskal–Wallis test between the three groups.
‡ Mann–Whitney U test between FH subjects Lp(a) ≥ 75 nmol/l and FH subjects Lp(a) < 75 nmol/l.
§ Mann–Whitney U test between FH subjects Lp(a) ≥ 75 nmol/l and controls.
║ Mann–Whitney U test between FH subjects Lp(a) < 75 nmol/l and controls.
Multivariable analyses
The results of multiple linear regression analyses of intake of n-6 PUFA in relation to plasma AA among FH subjects (n 139) were unchanged after adjustment for age, sex and BMI (crude regression coefficients: 0·32 (95 % CI 0·05, 0·60), P = 0 02 and adjusted regression coefficients: 0·32 (95 % CI 0·05, 0·59), P = 0·02, for every E % change in n-6 PUFA). Furthermore, the results of multiple linear regression analyses of intake of linoleic acid in relation to plasma AA among FH subjects (n 139) were unchanged after adjustment for age, sex and BMI (crude regression coefficients: 0·32 (95 % CI 0·05, 0·59), P = 0·02 and adjusted regression coefficients: 0·32 (95 % CI 0·05, 0·60), P = 0·02, for every E % change in linoleic acid). The results of multiple linear regression analyses of intake of AA in relation to plasma AA among FH subjects (n 139) were unchanged after adjusting for age, sex and BMI (crude regression coefficients: 4·9 (95 % CI −11·3, 21·1), P = 0·55 and adjusted regression coefficients: 0·79 (95 % CI −15·8, 17·3), P = 0·93, for every E % change in AA).
The results of the logistic regression analyses comparing plasma AA levels between the two FH groups were unchanged after adjusting for age, sex, BMI, use of PCSK9 inhibitors and cholesterol intake (crude and adjusted ORs were 1·29 (95 % CI 1·08, 1·54), P = 0·005 and 1·31 (95 % CI 1·08, 1·58), P = 0·006 for every unit change in plasma AA for FH subjects with elevated Lp(a) v. FH subjects without elevated Lp(a)).
Discussion
In the present study, we found that FH subjects with elevated Lp(a) levels had higher plasma levels of AA compared with FH subjects without elevated Lp(a). Furthermore, both FH subjects with and without elevated Lp(a) had higher plasma levels of AA compared with controls. Additionally, the associations between n-6 PUFA intake and plasma AA, and between plasma AA and Lp(a) in FH subjects, further support our hypothesis of a relationship between n-6 PUFA and Lp(a). We propose a novel link between n-6 PUFA intake, plasma AA and elevated Lp(a) levels.
AA and other higher n-6 PUFA derivatives are derived during desaturation and elongation processes from the precursor and essential FA linoleic acid(Reference Russo28); however, the plasma levels of AA may also depend on other processes such as turnover, oxidation and so on(Reference Hanna and Hafez29). Our results from both univariable and multivariable analyses suggest an association between plasma AA level and plasma Lp(a) level in FH subjects. Furthermore, the multivariable analyses show associations between plasma AA and n-6 PUFA intake in FH subjects, whereof dietary linoleic acid, but not AA, as expected, is the n-6 PUFA associated with plasma AA level. These results support that linoleic acid, which is the most abundant n-6 PUFA in the diet, is the driver of the association between dietary n-6 PUFA and plasma level of AA. Collectively, this may support an association between plasma Lp(a) concentration, dietary n-6 PUFA and plasma AA level. Consistent with our results, Hikita et al. have shown higher plasma Lp(a) concentration and lower ratio of EPA:AA in patients with compared with without increased risk of CVD(Reference Hikita, Shigeta and Kimura30). Recently, lipid apheresis was found to reduce linoleic acid and AA in the plasma of hyperlipidaemic patients, concomitant with a reduction in Lp(a) levels, further indicating an indirect link between n-6 PUFA and lipoproteins such as LDL and Lp(a)(Reference Schmocker, Kassner and Kiesler31,Reference Schmocker, Kassner and Ostermann32) . Previously, Li et al. showed that omnivores had both higher Lp(a) levels and higher concentration of AA in serum phospholipids(Reference Li, Ball and Bartlett33), possibly due to the increased direct availability of AA from their meat intake. However, the present study is, to our knowledge, the first to show a link between n-6 PUFA intake, plasma AA and plasma Lp(a) concentration in FH subjects.
The enzymes Δ-6 desaturase (D6D, rate limiting) and D5D convert linoleic acid to γ-linolenic acid and dihomo-γ-linolenic acid to AA, respectively, and the ratios 18 : 3n-6/18 : 2n-6 and 20 : 4n-6/20 : 3n-6 may be used as surrogate markers of D6D and D5D activities, respectively(Reference Kawashima, Sugawara and Okita25). Thus, our results may suggest higher D6D and D5D activity leading to lower plasma levels of linoleic acid and higher levels of downstream n-6 PUFA such as γ-linolenic acid, dihomo-γ-linolenic acid and AA in FH subjects compared with control subjects. Contrary, plasma levels of eicosadienoic acid were lower in FH subjects compared with controls. The conversion of linoleic acid to eicosadienoic acid is a ‘side track’ from the direct conversion towards AA(Reference Lee, Lee and Kang34), possibly indicating a preferred metabolism towards AA in FH subjects. Desaturases are regulated through the transcription factor, sterol regulatory element binding protein-1c, by dietary fat and cholesterol(Reference Nakamura and Nara35). Lower dietary intake of SFA and cholesterol may potentially be surrogate markers of higher PUFA intake and may thus differentially impact the regulation of D6D in FH subjects with elevated Lp(a). Thus, FA regulation of D6D may also partially explain the previously observed increased plasma levels of Lp(a) after lifestyle-induced, but not bariatric, weight reduction in the study by Berk et al. (Reference Berk, Yahya and Verhoeven4), further supporting the possible biological link between Lp(a) and AA. However, the link between n-6 PUFA and Lp(a) needs further investigation.
Common for the previously mentioned dietary RCT, the intervention groups lowered the intake of SFA but also increased the intake of carbohydrates(Reference Faghihnia, Tsimikas and Miller5–Reference Silaste, Rantala and Alfthan8), in particular the dietary fibre intake(Reference Berglund, Lefevre and Ginsberg6,Reference Silaste, Rantala and Alfthan8) . In line with this, we also find a higher intake of dietary fibre between the FH subjects with v. without elevated Lp(a). Hence, it cannot be ruled out that the diet-induced Lp(a) response may be a synergy effect of lowering SFA/increasing PUFA and increasing dietary fibre. In the present study, it is likely that all FH individuals received the same dietary advices regarding their diagnosis. However, the adherence to a beneficial diet is potentially better in the FH subjects with elevated Lp(a) concentration since they had experienced more events of CVD and were more often treated with PCSK9 inhibitors, potentially increasing the awareness of their increased CVD risk. Data on dietary pattern showed a lower intake of cakes, sweets and cheese in FH subjects with elevated Lp(a) compared with FH subjects without elevated Lp(a), indicating healthier food choices among FH subjects with elevated Lp(a). Although being mechanistically interesting, the diet-induced increase in plasma Lp(a) concentration is, however, probably less clinically significant since a reduction in approximately 100 mg/dl (approximately 240 nmol/l when converted according to Gencer et al.(Reference Gencer, Kronenberg and Stroes36)) in Lp(a) in a short-time perspective may be required to reduce the CVD risk with the same magnitude achieved by lowering LDL-cholesterol by 1 mmol/l(Reference Burgess, Ference and Staley37).
The FA content in phospholipids is dominated by n-6 PUFA, in particular linoleic acid and AA, rather than n-3 PUFA(Reference Spector38). Since Lp(a) transports more than 90 % of oxidised phospholipids (OXPL) in plasma, representing an atherogenic feature(Reference Tsimikas, Tsironis and Tselepis39), and there is an association between a dietary marker of increased PUFA intake and the proportion of different phospholipids(Reference Ruuth, Nguyen and Vihervaara40), it is tempting to speculate that increased levels of AA change the proportion of phospholipids, possibly increasing OXPL. Furthermore, linoleic and AA have been shown to be antagonists of farnesoid X receptor (FXR)(Reference Zhao, Yu and Lew41), and FXR activation has been shown to decrease Lp(a)(Reference Hoover-Plow and Huang42). Thus, n-6 PUFA may inhibit FXR, mediating an increase in Lp(a) required for transporting increased OXPL, and this small increase in Lp(a) could therefore represent a counteracting and even atheroprotective mechanism mediated by n-6 PUFA.
Major strengths of the study are that we show associations between plasma AA and Lp(a) concentrations in both univariable and multivariable analyses. Further, we included mainly genetically verified FH subjects with and without elevated Lp(a) with dietary data. A major limitation is the low number of control subjects. The main aim of the study was to compare FH subjects with and without elevated Lp(a) levels; however, we chose to include a small number of healthy controls to have a reference ‘point’ since a number of the analyses we measured (including the plasma FA profile) did not have established pre-specified cut-off points. Other limitations are that the study is explorative and hypothesis generating; the use of FA ratios as markers of enzyme activities since the plasma level of the product and the precursor FA may be biased by synthesis, turnover, oxidation, medication and so on; and measuring plasma FA as relative rather than absolute concentrations. However, a major scope of the study was to explore the relation between Lp(a) concentration and plasma levels of specific FA. Thus, we believe that relative values of FA are more meaningful in the present manuscript. Furthermore, FA in erythrocytes would have been a better marker for long-term diet intake reflecting FFQ data, compared with plasma FA from non-fasting blood samples that mainly reflect short-term dietary intake(Reference Hodson, Skeaff and Fielding43). Nevertheless, the FH patients had generally been followed up at the lipid clinic for years and received dietary counselling as part of their treatment. Furthermore, we have previously shown a more beneficial diet in children and young adults with FH compared with the general population(Reference Torvik, Narverud and Ottestad44). Also, we recently showed that 87 % of all FH adults, that had been treated at three Norwegian lipid clinics, received dietary counselling(Reference Bogsrud, Graesdal and Johansen45). Dietary pattern was measured using a score divided into three categories. LDL-cholesterol levels were lower among those with a diet score in the healthiest category at last visit than in subjects with a score in the most unhealthy category (3·2 (1·2) v. 4·4 (2·1) mmol/l, P < 0·001). After follow-up at the lipid clinics, the number of subjects with a diet score in the healthiest diet category doubled. These data are also supported by data from the SAFEHEART-study which showed that adults with FH have healthier dietary habits with lower consumption of SFA compared with non-FH(Reference Arroyo-Olivares, Alonso and Quintana-Navarro46). Taken together, this may support the notion that FH subjects have long-lasting improved dietary habits, which may be reflected in plasma FA even if plasma FA mainly reflect short-term dietary intake.
In conclusion, FH subjects with elevated plasma Lp(a) levels had higher plasma levels of the n-6 PUFA AA. Furthermore, dietary n-6 PUFA intake was associated with plasma AA, where dietary linoleic acid seemed to be the main driver of the dietary n-6 PUFA in this association. Our data suggest a novel link between plasma Lp(a) concentration, dietary n-6 FA and plasma AA concentration, which may contribute to explain the small diet-induced increase in Lp(a) levels associated with lifestyle changes. Although the increase may not be clinically relevant, this association may be mechanistically interesting in understanding more of the role and the regulation of Lp(a).
Acknowledgements
We gratefully thank all the subjects participating in this study. We also thank Navida Akther Sheikh and Anne Marte Wetting-Johansen at the Department of Nutrition, University of Oslo, Norway, and The Medical Laboratory at Oslo University Hospital, Rikshospitalet, for excellent technical assistance.
This study was supported by the University of Oslo, Oslo, Norway, the Norwegian National Advisory Unit on FH, Oslo University Hospital, Oslo, Norway, the Throne-Holst Foundation for Nutrition Research, Oslo, Norway and the South-Eastern Regional Health Authority, Oslo, Norway.
I. N., M. P. B. and K. B. H. conceived and designed research; I. N., M. P. B., L. K. L. Ø., T. U. and K. B. H. conducted research; I. N., L. K. L. Ø., M. B. V. and K. B. H. performed statistical analyses; I. N., M. P. B., L. K. L. Ø., S. M. U., K. R., M. M., J. R. v. L., B. H., P. A., M. B. V. and K. B. H., interpreted results; I. N., M. P. B., M. B. V. and K. B. H were responsible for drafting the manuscript; I. N., M. P. B., M. B. V. and K. B. H. were responsible for final content; all authors read, critically revised and approved the final manuscript.
M. P. B. received research grants and/or personal fees from Amgen, Sanofi, MSD, Boehringer Ingelheim, Mills DA and Kaneka, none of which are related to the content of this manuscript. K. R. received research grants and/or personal fees from Amgen, Mills DA, The Directorate for Health in Norway, The Norwegian Medical Association, Sanofi, Chiesi, Takeda, Bayer, MSD, none of which are related to the content of this manuscript. S. M. U. received research grants and/or personal fees from Mills DA, Tine BA and Rimfrost, none of which are related to the content of this manuscript. J. R. v. L. reports honoraria from Akcea and grants from Aegerion/Amryt, none of which are related to the content of this manuscript. K. B. H. reports grants and/or personal fees from Tine SA, Mills DA, Olympic Seafood, Amgen, Sanofi, Kaneka and Pronova, none of which are related to the content of this manuscript. The other authors have no financial relationships relevant to disclose.
Supplementary material
For supplementary material/s referred to in this article, please visit https://doi.org/10.1017/S0007114519001600







