


Flavone C-glucosides, important constituents of the flavonoid family, are mainly found in edible or medicinal plants, such as the tree Pterocarpus marsupium (Yadav & Singh, Reference Yadav and Singh1998), the fruits of Cucurbitaceae (Abou-Zaid et al. Reference Abou-Zaid, Lombardo, Kite, Grayer and Veitch2001) and the aerial parts of Gentianella species (Janković et al. Reference Janković, Krstić, Aljančić, Šavikin-Fodulović, Menković, Vajs and Milosavljević2005). They have diverse biological properties, including the antimicrobial activity of luteolin 6-C-glucoside (homoorientin) and apigenin 8-C-glucoside (vitexin) against Staphylococcus aureus, Bacillus subtilis and Pseudomonas aeruginosa (Afifi et al. Reference Afifi, Shervington and Darwish1997) and the protective effect of luteolin 7-O-glucoside against liver injury caused by carbon tetrachloride in rats (Qiusheng et al. Reference Qiusheng, Xiling, Xubo Meng and Changhai2004). Many researchers have investigated the remarkable antioxidant (Walle, Reference Walle2004) and radical scavenging activities (Salah et al. Reference Salah, Miller, Paganga, Tijburg, Bolwell and Rice-Evans1995) of the flavone C-glucosides, and also their inhibitory effects on LDL-C oxidation and lipid peroxidation (Kim et al. Reference Kim, Jun, Jeong and Chung2005), aiming to determine their protective effects on oxidative damage in both men and animals.
Coumaric acid belongs to the family of phenolic acids widely distributed in plants and forms part of the human diet (Scalbert & Williamson, Reference Scalbert and Williamson2000). Attempts have been made to explain the mechanisms of the antioxidant effects of phenolics. These include the binding of metal ions, scavenging of reactive oxygen species (ROS), reactive nitrogen species (RNS) or their precursors, up-regulation of endogenous antioxidant enzymes, or the repair of oxidative damage to biomolecules (Ursini et al. Reference Ursini, Tubaro, Rong and Sevanian1999). Epidemiological studies have suggested a statistical association between the consumption of phenolic acid-rich foods or beverages and the prevention of many diseases (Morton et al. Reference Morton, Caccetta, Ruddey and Croft2000). Recent interest in food-derived phenolic acids has increased because of their roles as antioxidants and scavengers of free radicals and their implication in the prevention of many pathological diseases such as cardiovascular diseases (Lowry et al. Reference Lowry, Rosenbrough, Farr and Randall1951; Ursini et al. Reference Ursini, Tubaro, Rong and Sevanian1999) and certain types of cancer (Hudson et al. Reference Hudson, Dinh, Kokubun, Simmonds and Gescher2000).
The increasing research has focused on the safety and metabolism of natural products, especially natural active components extracted from plants (Wolf & Weisbrode, Reference Wolf and Weisbrode2003). Bamboo is one of the most valuable natural plants worldwide, with nutrients extracted from its different edible parts (Rajebhosale et al. Reference Rajebhosale, Burte and Toro1998). In addition, published research data show different pharmacological activities on bamboo grass such as spontaneous motor activity (Nagasawa & Hatorri, Reference Nagasawa and Hatorri2001) and anti-tumour activity (Tsunoda et al. Reference Tsunoda, Yamamoto, Sakamoto, Inoue and Nagasawa1998). Bamboo is praised as the ‘Gold of the Poor’ because of its high economic value and it is as an important ingredient of forest resources. Antioxidant of bamboo leaves (AOB), a pale brown powder extracted from bamboo leaves, has been reported to be capable of blocking chain reactions of lipid auto-oxidation, chelating transient state metal ions, scavenging nitrite compounds and blocking the synthetic reaction of nitrosamine, as described in our previous study (Lou et al. Reference Lou, Zhang, Wu, Qi and Zhuo2004). However, other natural oxidants extracted from particular plants have innate deficiencies such as instability, low water solubility or an unpleasant odour so that their applications are restricted. For instance, the instability of tea polyphenol extract greatly affects its antioxidant use because auto-oxidation and epimerization are the two major reactions causing the instability of (–)-epigallocatechin-3-gallate (EGCG), the most abundant and biologically active compound in tea (Sang et al. Reference Sang, Lee, Hou, Ho and Yang2005). Compared to these extracts, AOB not only exerts a powerful antioxidant activity in various systems but also does not affect the sensory characteristics of the crude materials. To our knowledge, the active components extracted from bamboo leaves prepared in the present study play a significant role in the chemical, physiological and pharmacological utility of bamboo leaves. Moreover, the safety of the botanical extracts, AOB, is primarily ensured before the biological activities of AOB are studied and this problem has already been solved by our previous study (Lu et al. Reference Lu, Tie, Wu, Zhang and Zhang2005). Subsequently, AOB has been listed in the national standards (i.e. GB-2760) as a food antioxidant in China. In addition, permission has been granted authorized by the Ministry of Health, P.R. China (2003) to add AOB into extruded food, fish and meat products, and edible oils. The main functional components in AOB are flavonoids, lactones and phenolic acids. The flavone C-glucosides comprise a group of representative flavonoids in AOB, reported by Zhang et al. (Reference Zhang2005). The chemical structures of the four flavone C-glucosides, orientin, homoorientin, vitexin and isovitexin, found in AOB, are shown in Fig. 1. Like the phenolic acids in AOB, p-coumaric acid with a content of about 30 mg/kg in the presence of the free acid has been confirmed (Zhang, Reference Zhang2002).

Fig. 1 Chemical structures of the studied flavone C-glucosides in antioxidant of bamboo leaves (AOB). (A) orientin; (B) homoorientin; (C) vitexin; (D) isovitexin. Glu, glucose.
To explain the biological effects of both flavone C-glucosides and p-coumaric acid in AOB, it is assumed that they are bioavailable and effectively reach target tissues. A number of studies have been conducted on the metabolism of naturally occurring flavonoids, focused particularly on the role of the gut microflora in the metabolic transformation of flavonoids (Rice-Evans, Reference Rice-Evans2004; Whitman et al. Reference Whitman, Kurowska, Manthey and Daugherty2005). Few studies (Hattori et al. Reference Hattori, Shu, El-Sedawy and Namba1988) have been performed on the absorption, tissue distribution, metabolism and excretion (ADME) of flavone C-glucosides. In the present study, we identified and quantified the main metabolites absorbed in the blood and excreted in the urine and faeces of rats fed with an oral administration of AOB for 3 days. Quantitative analysis of flavone C-glucosides was performed by HPLC according to our previous study (Zhang et al. Reference Zhang, Bao, Lu, Ren, Tie and Zhang2005) and the ADME data for flavone C-glucosides and p-coumaric acid in AOB were reported systematically.
Materials and methods
Preparation of AOB
AOB was provided by Zhejiang University Innoessen Co., Ltd (Hangzhou, China). It was prepared from the bamboo leaves of Phyllostachys nigra var. henonis identified by the Research Institute of Subtropical Forestry of the Chinese Academy of Forestry, Hangzhou, China (Zhang et al. Reference Zhang, Wu, Ren, Fu and Zhang2004). In brief, fresh bamboo leaves were collected during the autumn season in Anji district (Zhejiang Province, China) and air dried. The coarse powder of the bamboo leaves was obtained by crushing into 20–40 mesh and 10 g powder was extracted during 1 h with 100 ml 30 % (v/v) aqueous ethanol solution using the hot reflux method. The filtrate was then isolated by membrane filtration to remove macro- and micro-molecular components such as polysaccharides and minerals. AOB was finally obtained after concentrating under vacuum and spray drying. The full sample analysis data for AOB including the flavonoid, lactone and phenolic acid monomer content were reported previously (Lu et al. Reference Lu, Tie, Wu, Zhang and Zhang2005).
Chemical reagents and instrumentation
Four flavone C-glucoside standards, orientin (3′,4′,5,7-tetrahydroxyflavone-8-C-glucoside), homoorientin (3′,4′,5,7-tetrahydroxyflavone-6-C-glucoside), vitexin (4′,5,7-trihydroxyflavone-8-C-glucoside) and isovitexin (4′,5,7-trihydroxyflavone-6-C-glucoside) standards (HPLC-grade) were obtained from Extrasynthese Company Inc. (Lyon, France). Hydrocaffeic acid (HCA), phloretic acid (PA), phloroglucinol (PG) and p-coumaric acid was purchased from Sigma-Aldrich Co. (St Louis, USA). Acetonitrile (HPLC-grade) was provided by Tedia Company Inc. (OH, USA) and methanol was purchased from Merck & Co., Inc. (NJ, USA). Orientin, homoorientin, vitexin, isovitexin, HCA, PA, PG and p-coumaric acid have been previously confirmed by MS and 1H and 13C NMR (Pouchert & Behnke, Reference Pouchert and Behnke1993). All of the flavonoids and p-coumaric acid were checked for purity, prior to use, by HPLC and were found to be >98 % pure. In particular, it was necessary to ensure that the corresponding aglycones of four flavone C-glucosides (luteolin for orientin/homoorientin and apigenin for vitexin/isovitexin) were not detected. Otherwise the impurity of these flavone C-glucosides would interfere with the results of the metabolism and transport studies. Heparin sodium (6·250 U/ml), used as the anticoagulant, was provided by Ningbo Biochemistry Reagents Co. (Ningbo, China). All other chemical reagents were of analytical grade. Water was purified with the Milli-Q system (Millipore, Bedford, USA). All the solutions prepared for HPLC were passed through a 0·45 μm nylon filter before use.
The chromatographic system used for the quantitative analysis of the metabolites consisted of an HP-1100 series high-performance liquid chromatograph from Agilent Technologies (Palo Alto, CA, USA) equipped with a quaternary pump, on-line degasser, autosampler, automatic injector, column heater and diode-array detectors connected on-line. The metabolites were quantified with a Luna C18 column (5 μm, 250 mm × 4·6 mm i.d.) protected by an RP18 guard column (5 μm, 4·0 mm × 3·0 mm i.d.), both from Phenomenex (Torrance, CA, USA). The identification of p-coumaric acid and degradation products from flavone C-glucosides was performed on a Hewlett-Packard (HP) 6890 gas chromatograph (GC) coupled to an HP 5973 benchtop mass selective detector (MSD) operated in selected ion monitoring (SIM) mode with positive electron impact (EI) ionization. The chromatographic elution was performed on an HP5-MS capillary column (polysiloxane polymers, 30 m × 0·25 mm, 0·25 μm, J&W Scientific, Agilent, CA, USA). A vortex mixer WH-861 Minishaker (Taicang Science & Educational Instrumental Co., Jiangsu, China) was used to mix and homogenize the biological samples during pretreatment. An ultracentrifuge with a cooling system (Beckman J-20-XP Series, Krefeld, Germany) was used for protein removal from the sample. The pH of the buffer solutions and samples was adjusted with a PHS-3C precision pH meter (Leici Instrumental Co., Shanghai, China). Solid-phase extraction was carried out on a VacElut vacuum manifold for 20 cartridges (Varian, Palo Alto, CA, USA).
Animals, diets and oral administration
Twenty male and twenty female Sprague-Dawley (SD) rats (Shanghai Laboratory Animal Centre, Shanghai, China) weighing 180–190 g at the beginning of the experiment were housed, two per feeding cage, in a temperature and relative humidity controlled room (22–25°C, 40–60 %), which was maintained in an inverse 12-h light/dark cycle. After 2 weeks of adaptation in this experimental environment, thirty-six of the rats were divided randomly into nine groups (Groups A–I, two males and two females for each group) for the absorption and tissue distribution studies. The remaining four rats (Group J) were used in the pharmacokinetics and excretion study. Animals were handled according to China National Standards (GB14924-1994) and received a basal diet, comprising casein (1·0 %), soy bean powder (15·0 %), wheat flour (54·0 %), corn oil (4·0 %), cellulose (2·0 %), mixed salts (2·6 %), mixed vitamins (1·0 %), choline chloride (0·2 %), methionine (0·2 %) and starch (11·0 %), supplied by the Zhejiang Laboratory Animal Centre (Hangzhou, China), during adaptation periods and were fasted overnight, but water was available ad libitum before the ADME study. In the preliminary test, two male and two female rats fed with the basal diet were fasted overnight and killed by decollated exsanguination the next day. All of the biological samples related to the ADME study, which included blood, gastrointestinal organs, liver, brain, kidneys, thigh muscle tissues, faeces and urine, were carefully collected and analysed by HPLC after routine pretreatment optimized in the present study. The results showed that no primary parent compounds or metabolites studied in the present work were detected, and indicated that overnight fasting was sufficient to ensure that the basal diet would not interfere with the ADME study.
When the ADME study of AOB was performed, rats were fed by gavage; the AOB (1 g/kg body weight) was dissolved in water (1 g AOB per 10 ml) containing four flavone C-glucosides and p-coumaric acid. The oral administration level of AOB was designed according to the no-observed-adverse effect level (NOAEL) of AOB determined by an acute and subchronic toxicity study (Lu et al. Reference Lu, Tie, Wu, Zhang and Zhang2005). All of the animals tested received a single-time oral gavage of an aqueous suspension of AOB at the above-mentioned level. Biological samples including blood, urine, faeces and viscera were collected at corresponding time points and stored immediately at − 20°C until analysis.
Chromatographic analysis
A modified version of an analytical HPLC method described previously (Zhang et al. Reference Zhang, Bao, Lu, Ren, Tie and Zhang2005) was used for the quantitative analysis of the metabolites. Solvent A (99/1 (v/v) water/acetic acid) adjusted to pH 3·0 with NaOH and solvent B (acetonitrile) were run at a flow rate of 1 ml/min. A gradient programme was used as follows: 15 % B (15 min), 15–40 % B (10 min), 40 % B (9 min), 40–15 % B (6 min). A column clean-up stage was performed at 100 % B (5 min) followed by a re-equilibration at 15 % B (15 min). The column was packed with Luna 5 μm C18 reversed-phase silica, 250 × 4·6 mm i.d. (Phenomenex). Aliquots of 30 μl were injected directly into the HPLC system and the eluent was monitored at 330 nm with a diode-array detector.
For the chemical identification by GC-MS, the GC was operated at a constant flow of helium with a flow rate of 1·0 ml/min. The injector temperature was set at 300°C and 1 μl of sample was injected in the splitless mode. The initial oven temperature was set to 150°C for 3 min, followed by a programmed temperature ramp of 15°C/min to 280°C, and the temperature was held at this level for 20 min. The MSD was set to acquire data in the full scan mode (mass range 50–550) at 2·9 scans/s after 5 min of solvent delay time, with a multiplier voltage of 1388 V and ionization energy of 70 eV. The MS source and interface temperatures were 230°C and 280°C, respectively.
ADME study of AOB
After dosing, animals were maintained in wire cages and those from Groups A–I were killed by decollated exsanguination at their corresponding time points, that is four rats for each time point at 0.5, 1, 1.5, 2, 3, 4, 6, 8 and 12 h. The biological samples, including brain, liver, kidneys and thigh muscle tissues, were carefully collected at the corresponding time points and stored immediately at − 20°C until the tissue distribution analysis of analytes, including flavone C-glucosides and p-coumaric acid. The main organs of the gastrointestinal tract (the stomach, intestines and colon) were also carefully taken at corresponding time points for the gastrointestinal tract absorption analysis of analytes and immediately stored at − 20°C. To determine the pharmacokinetics of the analytes, blood samples (approximately 150 μl) were taken from the lateral caudal vein of rats in Group J at the above-mentioned time points, collected in heparinized microhaematocrit tubes and stored in a refrigerator until analysis.
For the excretion study, rats from Group J were placed in individual all-glass metabolism cages for the collection of urine and faeces after dosing. Accumulated urine and faeces were collected at 12, 24, 36, 48, 60 and 72 h, that is at 12-h intervals up to 72 h after dosing. As for each sampling, cage debris was collected and the collecting funnels were rinsed with water. At the end of the collection period, the cages were rinsed with water followed by methanol. All cage rinses were retained for analysis.
When the pretreatment of the biological samples was performed, aliquots (100 μl) of blood samples were transferred into centrifuge tubes. After adding 200 μl of methanol, the samples were shaken on an ultrasonic vibrator for 30 min to adequately extract the analytes and then 100 μl of water was added to adjust the ratio of methanol/water (1:1, v/v) because preliminary assays suggested that the optimal level of methanol as dilution medium was 40–50 % (v/v) according to total response areas of the four flavone C-glucosides (for details, see Discussion). Samples were allowed to stand at 4°C for several minutes and then centrifuged at 48 520 g for 10 min (4°C). An aliquot (30 μl) of the supernatant was injected directly into the HPLC. Similar pretreatment procedures of other biological samples collected were performed and the sampling aliquots were determined as about 1·5–2 g for different samples and 0·5 ml for the urine sample.
To determine the metabolites of AOB, particularly aromatic acids in the excretion samples, urine samples and the methanol extracting solution of faeces (200 μl each) were acidified to pH 5 with 20 μl of 0·6 mol/dm3 acetic acid and incubated at 37°C for 30 min in the presence of 1100 U β-glucuronidase and 42 U sulfatase (Helix pomatia extract, Sigma Chemical Co.). After further acidification to pH 2 with HCl 6 mol/dm3, the urine was extracted twice with ethyl acetate and centrifuged at 1090 g for 10 min. The resulting supernatant was dried under nitrogen, redissolved in 500 μl of methanol, and filtered (PTFE membrane, 0·45 μm, Millipore, Bedford, MA, USA). An aliquot (1 μl) of the filtrate was injected directly into the GC-MS system. Aromatic acids and other metabolites were detected according to the respective m/z values of their molecular ions and fragment ions.
Bioavailability estimation of p-coumaric acid in rats
To investigate the metabolic behaviour and calculate pharmacokinetic parameters, the bioavailability of p-coumaric acid was estimated by comparison of the areas under the concentration–time curves (AUC) after intravenous and oral administration in rats. The animal grouping and the experimental design of the animal test were described earlier in the ADME study. Rats were orally administrated or intravenously injected with 100 μmol/kg body weight of p-coumaric acid. Three rats were assigned to each time point of each experimental group.
Calculations and statistics
Two-compartmental pharmacokinetic evaluations of blood data were performed according to the corresponding compartment model theory (Green et al. Reference Green, Swain, van Miller and Joiner2003). The pharmacokinetic parameters were calculated based on the functional expression of the two-compartment model deduced by the residual theorem. The AUC was calculated by using the definite and improper integral rule. AUC(0–t) was defined as the area under the curve between time zero and the last datum point. AUC(0–∞) was extrapolated from the last datum point using the rate constant defined in the terminal elimination phase. Routine parameters including apparent volume of distribution (V), total body clearance (Cl), rate constant (k) and half-life (t 1/2) were calculated. Comparisons of results were performed by ANOVA. Numerical values were expressed as means and standard deviations.
Results
Excretion study of AOB
In the urine of oral administrated SD rats (Group J), p-coumaric acid was detected and found to be excreted as its original analyte but cumulative excretions of p-coumaric acid at 12 and 24 h were only 1·80 (sd 0·24) % and 1·90 (sd 0·26) % of the dose, respectively, and after 24 h no analyte could be detected. However, not all of the flavone C-glucosides were detected in the HPLC chromatogram of urine samples during the whole metabolic period (Fig. 2(a)). Conversely, p-coumaric acid could not be detected in the HPLC chromatogram of faeces samples from oral administrated SD rats (Fig. 2(b)) during the whole metabolic period, while the four flavone C-glucosides were detected and determined in the excretions as their parent forms, and cumulative excretions of orientin, homoorientin, vitexin, isovitexin and total amounts at 24 h were 24·04 (sd 2·17), 24·05 (sd 1·91), 9·72 (sd 0·88), 20·97 (sd 1·90) and 21·23 (sd 1·92) % of the dose, respectively (n 4), and declined rapidly with time. However, the four flavone C-glucosides at the other time points (12, 36, 48, 60 and 72 h) were not detected.
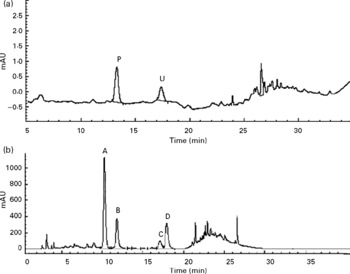
Fig. 2 HPLC chromatograms of (a) urine sample and (b) faeces samples during the whole metabolic period of the excretion study of antioxidant of bamboo leaves (AOB) in rats. (A) orientin; (B) homoorientin; (C) vitexin; (D) isovitexin; (P) p-coumaric acid; (U) unknown peak.
Tissue distribution study of AOB
Neither the flavone C-glucosides nor p-coumaric acid were detected in the brain, liver and thigh muscle tissues within 12 h at any time point. However, p-coumaric acid was detected in the kidneys and was observed to increase and decrease at different time points (Fig. 3), reaching a peak value of 14·26 μg/g at 1.5 h. The bioavailability of p-coumaric acid was estimated as approximately 74 % in the rats. Data on the elimination of p-coumaric acid in the urine of rats could then be calculated as the difference between the maximum level of p-coumaric acid in the kidneys and the residual level at different time points. Based on these data, the accumulated elimination level of p-coumaric acid in urine (X u) at different time points (t) could be summarized by Equation (1), which was fitted according to the least squares approximation:
Corresponding urine elimination parameters were calculated as follows: first-order rate constant of elimination k = 0·55 1/h; rate constant of kidney elimination k e = 0·002 1/h; half-life of elimination phase t 1/2 = 1.25 h; kidney clearance Clr = 1·61 l/h.
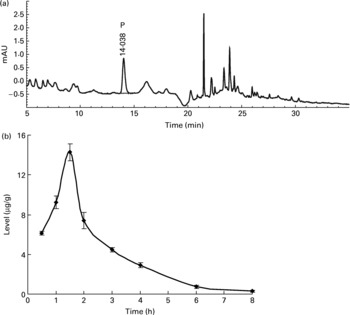
Fig. 3 (a) The HPLC chromatogram of the extract from kidneys during the whole metabolic period of the tissue distribution study of antioxidant of bamboo leaves (AOB) in rats and (b) the concentration of p-coumaric acid (♦) at different time points (n 4). P, p-coumaric acid. Error bars correspond to standard deviations.
Gastrointestinal tract elimination of AOB and metabolic pathway of flavone C-glucosides
The rats orally fed AOB showed a positive correlation between the elapsed time and the decrease in flavone C-glucosides. During the whole metabolic process, the recovery of total flavone C-glucosides was found by examination of gastrointestinal tract extracts in each time point (Groups A–I) to be 83·4 (sd 0·8), 68·8 (sd 1·3), 65·1 (sd 1·5), 61·0 (sd 2·9), 59·8 (sd 3·3), 59·1 (sd 2·0), 57·8 (sd 3·2), 53·7 (sd 1·1) and 51·7 (sd 0·8) % at 0.5, 1, 1.5, 2, 3, 4, 6, 8 and 12 h, respectively (n 4, Fig. 4(d)). The data indicated that more than 50 % of the flavone C-glucosides were metabolized and excreted as their parent compounds (Fig. 4(a,b)), and were not involved in kidney metabolism (Fig. 4(c)). In other words, these four flavone C-glucosides could not enter the liver through the portal vein and be involved in the enterohepatic cycle. Therefore, the metabolic process of flavone C-glucosides can be summarized by Equation (2):
where M 1 and M 2 are the amounts of flavone C-glucosides detected in gastrointestinal tract extracts and faeces, respectively, and M 3 is the amount of flavone C-glucosides that were hydrolysed by gut microflora or conjugated by unknown pathways. ΔM refers to the evaluated error derived from all of the above three factors.
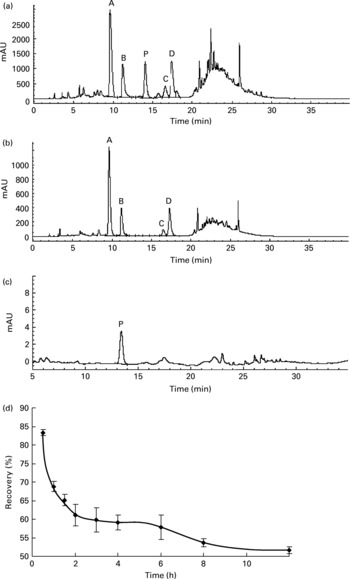
Fig. 4 HPLC chromatograms of the extract from (a) the stomach, (b) the small intestine and (c) blood during the gastrointestinal tract elimination study of antioxidant of bamboo leaves (AOB), and (d) the gastrointestinal tract elimination growth and decline curve of total flavone C-glucosides in rats. (A) orientin; (B) homoorientin; (C) vitexin; (D) isovitexin; (P) p-coumaric acid; (♦), the recovery of the four flavone C-glucosides at each time point. Error bars correspond to standard deviations.
Identification of degradation products from flavone C-glucosides
Besides the metabolism of the flavone C-glucosides in the original sample, the metabolic pathway of the hydrolysation of the analytes by gut microflora was demonstrated. The residues extracted from the gastrointestinal tract and faeces were extracted by ethyl acetate, purified by solid-phase extraction and analysed by the GC-MS system. Some metabolites were detected by GC-MS and their structures were identified and confirmed. PG, HCA and PA were also detected, and their mass spectra were confirmed by spectra from GC-MS databases. We also confirmed the peaks identified as PG, HCA and PA from the results, in which the retention times of the peaks agreed with those of correlative standard reagents for GC-MS analysis. The concentrations of the three metabolites PG, HCA and PA in the gastrointestinal tract extracts and faeces of rats at different time points were detected, as shown in Table 1. By comparison of the chemical structures of flavone C-glucosides and phenols, the metabolic pathway of the flavone C-glucosides hydrolysed by the gut microflora can be summarized as deglycosylation and ring-opening of the heterocyclic C ring. The proposed metabolic pathways of orientin, vitexin and isovitexin are shown in Fig. 5. As an example, homoorientin was hydrolysed to its aglycone luteolin, or hydrogenated into 3′,4′,5,7-tetrahydroxy-flavanone-6-glucoside, which was then deglycosylated into ( ± )-eriodictyol. This was followed by ring-opening of the heterocyclic C ring, generating PG and HCA.
Table 1 Phloroglucinol (PA), hydrocaffeic acid (HCA) and phloretic acid (PA) levels in the gastrointestinal tract extracts and faeces of rats at different time points*
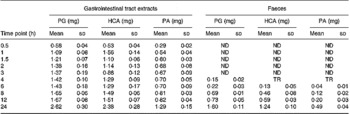
ND, not detected; TR, trace level but could not quantify.
* Each value represents the mean for four different samples (n 4).
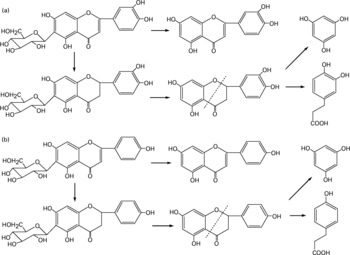
Fig. 5 Proposed metabolic pathways of (a) homoorientin and (b) isovitexin degradation initiated by the gut microflora. The metabolic pathways of orientin and vitexin were similar to homoorientin and isovitexin, respectively.
Identification and pharmacokinetics of p-coumaric acid
To further confirm whether p-coumaric acid was absorbed in the gut in its original form, a biological sample was determined by HPLC and GC-MS and compared to p-coumaric acid extracted directly from AOB. In brief, p-coumaric acid was extracted twice from AOB with ethyl acetate and ether, and then purified by C18 cartridge solid-phase extraction using 15 % (v/v) methanol as eluent. Figure 6 shows the HPLC and GC-MS chromatograms of p-coumaric acid extracted directly from AOB. Similarly, the mass spectrum of p-coumaric acid was in correspondence with the spectrum from the GC-MS databases. The purified p-coumaric acid prepared from AOB was also submitted for structural identification by IR, UV, MS and NMR, and the corresponding data were similar to data published previously (Pouchert & Behnke, Reference Pouchert and Behnke1993). The results of the pharmacokinetic study showed that the amount of p-coumaric acid in blood (17·25 μg/ml) reached a peak value at 1 h. Figure 7 shows the pharmacokinetic curve of p-coumaric acid in vivo. Based on the two-compartment model theory, there is a central compartment (tissues and organs with abundant blood flow and instantaneous distribution, such as heart, liver and kidneys) and a peripheral compartment (tissues and organs with little blood supply and slow analyte distribution, such as skeleton, fats and muscle). The whole pharmacokinetic process can described by Equation (3):
Corresponding pharmacokinetic parameters were calculated as followings: first-order rate constant of transportation from central to peripheral compartment k 12 = 0.52/h, transportation from peripheral to central compartment k 21 = 0.55/h and elimination from central compartment k 10 = 0.60/h; half-life of absorption phase t 1/2(a) = 0.11 h, distribution phase t 1/2(α) = 0.48 h and elimination phase t 1/2(β) = 3.02 h; AUC(0–∞) = 25·81 μg h/ml and AUC(0–t) = 23·20 μg h/ml; apparent volume of distribution V = 767·07 l; total body clearance Cl = 175·95 l/h.
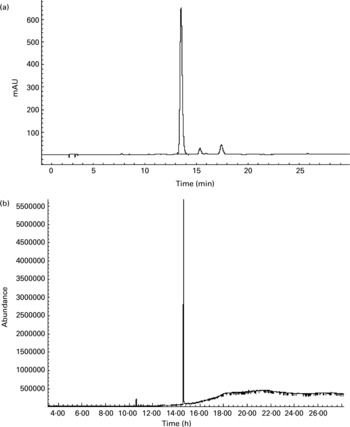
Fig. 6 (a) HPLC and (b) GC-MS chromatograms of p-coumaric acid extracted directly from antioxidant of bamboo leaves (AOB). The retention times of p-coumaric acid were 13.5 and 14.6 min in HPLC and GC-MS chromatograms, respectively.
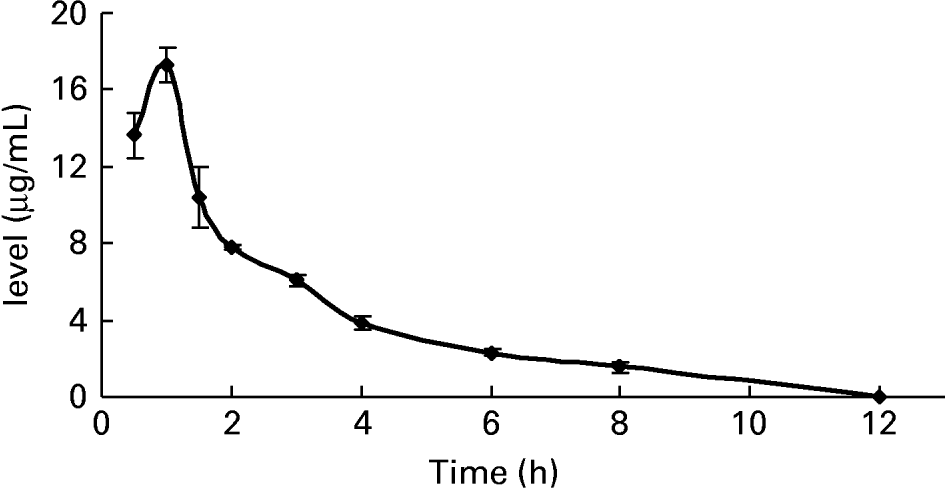
Fig. 7 The pharmacokinetic curve of p-coumaric acid in blood of rats at different time points after oral administration of antioxidant of bamboo leaves (AOB) (n 4). Error bars corresponded to standard deviations.
Discussion
The antioxidant properties and relative biological responses of AOB have been investigated in depth in our previous studies. Zhang et al. (Reference Zhang, Tang and Ding1998) systematically investigated the bio-antioxidant properties of AOB both in vitro and in vivo. AOB was shown to effectively scavenge superoxide anion radicals ( ) produced by the xanthine–xanthine oxidase–luminol chemiluminescence system (Banu et al. Reference Banu, Greenway and Wheatley2005) in the concentration range 0·1–5000 mg/l (IC50 = 0·8 mg/l), and radicals were completely eliminated when the concentration of AOB was in the range 10–5000 mg/l. AOB was also able to scavenge hydroxyl radicals (√OH) produced by the vitamin C–Cu2+–H2O2–yeast polysaccharide–luminol chemiluminescence system (Tsukagoshi et al. Reference Tsukagoshi, Nakahama and Nakajima2003) within the concentration range 0·01–5000 mg/l (IC50 = 320 mg/l). The strong scavenging activity of AOB was confirmed by pyrogallol auto-oxidation and electron spin resonance (ESR) methods. Results of a feeding test in vivo showed that AOB could significantly improve the activity of superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px) measured by chemiluminescence and 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) methods in aged mice, especially when the dose levels of AOB were set at 0· 5g/kg and 1·0 body weight per day, respectively (P < 0·05, P < 0·001; Zhang et al. Reference Zhang, Tang and Ding1998). Hu et al. (Reference Hu, Zhang and Kitts2000) further demonstrated the antioxidant activity of AOB and its biological responses, reporting that AOB exhibited a good concentration-dependent scavenging activity of the 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical, prolonged the lag phase and reduced the rate of propagation of liposome peroxidation initiated by the peroxyl radical induced by 2,2′-azobis(2-amidinopropane dihydrochloride) (AAPH), prevented human LDL oxidation, mediated by Cu2+, and protected supercoiled DNA strands against scission induced by the AAPH-mediated peroxyl radical. Based on all of these antioxidant-related studies in vitro and in vivo, AOB can be regarded as an effective natural antioxidant. In the present study, the metabolic processes of flavone C-glucosides and p-coumaric acid, the main active components of AOB, are discussed systematically and the data obtained may be used in explaining the mechanisms of the physiological and pharmacological activities, including the antioxidant activity, of this botanic extract.
) produced by the xanthine–xanthine oxidase–luminol chemiluminescence system (Banu et al. Reference Banu, Greenway and Wheatley2005) in the concentration range 0·1–5000 mg/l (IC50 = 0·8 mg/l), and radicals were completely eliminated when the concentration of AOB was in the range 10–5000 mg/l. AOB was also able to scavenge hydroxyl radicals (√OH) produced by the vitamin C–Cu2+–H2O2–yeast polysaccharide–luminol chemiluminescence system (Tsukagoshi et al. Reference Tsukagoshi, Nakahama and Nakajima2003) within the concentration range 0·01–5000 mg/l (IC50 = 320 mg/l). The strong scavenging activity of AOB was confirmed by pyrogallol auto-oxidation and electron spin resonance (ESR) methods. Results of a feeding test in vivo showed that AOB could significantly improve the activity of superoxide dismutase (SOD) and glutathione peroxidase (GSH-Px) measured by chemiluminescence and 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) methods in aged mice, especially when the dose levels of AOB were set at 0· 5g/kg and 1·0 body weight per day, respectively (P < 0·05, P < 0·001; Zhang et al. Reference Zhang, Tang and Ding1998). Hu et al. (Reference Hu, Zhang and Kitts2000) further demonstrated the antioxidant activity of AOB and its biological responses, reporting that AOB exhibited a good concentration-dependent scavenging activity of the 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical, prolonged the lag phase and reduced the rate of propagation of liposome peroxidation initiated by the peroxyl radical induced by 2,2′-azobis(2-amidinopropane dihydrochloride) (AAPH), prevented human LDL oxidation, mediated by Cu2+, and protected supercoiled DNA strands against scission induced by the AAPH-mediated peroxyl radical. Based on all of these antioxidant-related studies in vitro and in vivo, AOB can be regarded as an effective natural antioxidant. In the present study, the metabolic processes of flavone C-glucosides and p-coumaric acid, the main active components of AOB, are discussed systematically and the data obtained may be used in explaining the mechanisms of the physiological and pharmacological activities, including the antioxidant activity, of this botanic extract.
Flavone C-glucosides are poorly absorbed in the gut because of the direct linkage between the flavone aglycone and the corresponding glucoside, not because of the oxo bridge and their high molecular weight. Flavone C-glucosides largely reach the colon where they can be degraded into various aromatic acid metabolites by the microflora. These microbial metabolites are formed by cleavage of the A ring from the residual flavone C-glucoside molecule and the opening of the heterocyclic C ring and could also contribute to their biological activity (Aherne & O'Brien, Reference Aherne and O'Brien2002). Little is known about the abundance of flavone C-glucosides in vivo. In the present work, we examined the metabolic fate of flavone C-glucosides in rats, and evaluated the absorption, tissue distribution and excretion of flavone C-glucosides in AOB in relation to their metabolism for the first time. For the flavone O-glucosides, deglycosylation has been proposed as the first stage of metabolism. Some are hydrolysed to their aglycones by the small intestine and liver enzymes. After absorption, flavone O-glucosides are bound to albumin and transported to the liver through the portal vein (Manach et al. Reference Manach, Regerat, Texier, Agullo, Demigne and Remesy1996). Finally, they reach the blood circulatory system and enter the enterohepatic cycle. However, conjugation reactions with glucuronic acid and/or sulfate also seem to be the most common type of metabolic pathway reported for the flavone O-glucosides (Rice-Evans, Reference Rice-Evans2004; Walle, Reference Walle2004). In the present study, the flavone C-glucosides homoorientin, orientin, isovitexin and vitexin are not readily absorbed in the gut. Conversely, they directly reached the colon where they are hydrolysed into their aglycones, luteolin and apigenin, and degraded into small-molecule phenols and various aromatic acids, as in the metabolism of dietary procyanidins reported by Gonthier et al. (Reference Gonthier, Donovan, Texier, Felgines, Remesy and Scalbert2003). Therefore, the antioxidant activity of these four flavone C-glucosides requires consideration of both the parent compounds and the metabolites. For instance, the homoorientin and orientin heterocycle contribute to antioxidant activity by the presence of 3′,4′-catechol, and flavonoids with a 3′,4′-catechol have been reported to be ten times more potent than ebselen, a well-known RNS scavenger, against peroxynitrite (Heijnen et al. Reference Heijnen, Haenen, Vekemans and Bast2001). In the present study, the rate of elimination of the four flavone C-glucosides from the gastrointestinal tract was low, even though 50 % recovery of flavone C-glucosides was determined at 12 h in the gastrointestinal tract (Fig. 4(d)) and faeces containing these four analytes were excreted at 24 h. These data suggest that the effective time of analytes in the colon was long enough so that they could exert their antioxidant activity and scavenge free radicals.
Additionally, some small polyphenol molecules such as PG, HCA and PA were also detected in the gastrointestinal tract extracts and faeces of rats at different time points (Table 1). It was observed that the PG level in both gastrointestinal extracts and faeces was mostly higher than other two phenols, and also the content of the metabolite HCA was higher than that of PA. The regularity of different contents of three polyphenol molecules could partly demonstrate the metabolic pathways of flavone C-glucoside degradation initiated by the gut microflora. Based on their chemical structure, it is likely that HCA was derived from homoorientin and oritentin while PA was derived from isovitexin and vitexin. The difference in concentration of the three compounds could be clearly elucidated according to the different contents of the four flavone C-glucosides in AOB. As shown in Fig. 5, the initial step is hydrogenation of the flavone C-glucosides due to the position of the 2,3-double bond on the C-ring. The aglycone intermediates are the main precursors of above-mentioned polyphenols, which were formed after the deglycosylation.
In this study, p-coumaric acid was absorbed well in the gut because of its low molecular weight, and the bioavailability of p-coumaric acid was relatively high, at approximately 74 %, in agreement with other published reports. Konishi et al. (Reference Konishi, Hitomi, Yoshida and Yoshioka2005) estimated the bioavailability of p-coumaric acid, as the ratio of the AUC in the abdominal artery to the area under the plasma concentration time curve (AUC) in the portal vein, to be about 0·70. Compared to p-coumaric acid from blood and urine, it could be confirmed that p-coumaric acid was absorbed and excreted in its original form. p-Coumaric acid has reported antioxidant effects such as up-regulation of endogenous antioxidant enzymes and the repair of oxidative damage to biomolecules so that it is recognized as an oxidative inhibitor (Ursini et al. Reference Ursini, Tubaro, Rong and Sevanian1999). Abdel-Wahab et al. (Reference Abdel-Wahab, El-Mahdy, Abd-Ellah, Helal, Khalifa and Hamada2003) proved the beneficial effect of the natural phenolic acid p-coumaric acid in protecting animals against doxorubicin-induced cardiac oxidative damage and suggested that this protecting potential of p-coumaric acid could be due to its free radical-scavenging ability. On the basis of their findings, it may be worthwhile suggesting the concomitant administration of p-coumaric acid with doxorubicin cancer chemotherapy.
Understanding the biodynamics of flavone C-glucosides and p-coumaric acid after oral administration is fundamental to understanding the existing structure–activity relationship and its potential use in preventive nutrition. In addition to structural and physico-chemical attributes of the nascent compound, the absorption, pharmacokinetics and biotransformation to certain metabolites are crucial determinants of biological effects in organisms. Data obtained both in vitro and in vivo effectively and consistently demonstrate the antioxidant efficacy of structurally diverse flavonoids and aromatic acids under many circumstances of oxidative stress. However, the current understanding of absorption and metabolism is limited to a small number of dietary flavonoids and few studies refer to the metabolism of flavone C-glucosides (Heim et al. Reference Heim, Tagliaferro and Bobilya2002). To integrate new knowledge of structure–activity relationship into the areas of human nutrition and medicine, future research should investigate (i) the extent of absorption in relation to structure, (ii) pharmacokinetics in human subjects, (iii) characterization of the metabolites, (iv) the structure–activity relationship and health effects of these metabolites, and (v) the metabolic fate of other phenolic acids and their competitive absorption mechanisms in vivo.
Acknowledgements
This work was supported by grants-in-aid from the National Natural Science Foundation of China (No. 30540016).








