


Dietary fat is a major energy source except providing essential fatty acids (FA) that are important for health. However, alterations in the amount and composition of dietary fat, especially an increase of n-6:n-3 PUFA ratio, may contribute to the development of obesity( Reference Allhaud, Guesnet and Cunnane 1 ). In the recent decades, increasing attention has been given to the role of the small intestine in the development of high-fat diet (HF)-induced obesity, as a high level of FA could affect intestinal lipid metabolism( Reference de Wit, Bosch-Vermeulen and de Groot 2 ). The small intestine is the gatekeeper at the physical interface between the body and the diet( Reference de Wit, Bosch-Vermeulen and de Groot 2 ) and one of the main organs expressing β-oxidation-related enzymes( Reference Kelly, Gordon and Alpers 3 , Reference Nemali, Usuda and Reddy 4 ). As a result, lipid catabolism and uptake in the intestine may have a critical role in the aetiology of obesity.
Recently, dietary n-3 PUFA has been suggested to increase intestinal expression of genes related to FA oxidation( Reference van Schothorst, Flachs and Franssen-van Hal 5 , Reference Mori, Kondo and Hase 6 ) and cholesterol uptake( Reference van Schothorst, Flachs and Franssen-van Hal 5 ). However, the mechanism demonstrating how n-3 PUFA modulates lipid metabolism in the intestine remains to be elucidated. Many studies demonstrate that n-3 PUFA exert their effects on lipid metabolism through the PPAR pathway( Reference van Schothorst, Flachs and Franssen-van Hal 5 , Reference Kimura, Takahashi and Lin 7 ), as n-3 PUFA are intrinsic ligands for PPAR( Reference Keller, Dreyer and Medin 8 ). Moreover, n-3 PUFA could interact with PPARα and enhance PPARα transcriptional activity( Reference Keller, Mahfoudi and Dreyer 9 ). However, other studies report that expression of carnitine palmitoyltransferase 1 (Cpt1), a key gene involved in FA oxidation, could be induced by FA in the absence of PPARα ( Reference Louet, Chatelain and Decaux 10 , Reference Le May, Cauzac and Diradourian 11 ). These results suggest that other mechanisms independent of PPAR are involved in n-3 PUFA-induced FA catabolism.
AMP-activated protein kinase (AMPK) is the key sensor of whole-body energy homeostasis, and it has a critical role in the regulation of lipid metabolism through modulating Cpt1( Reference Ronnett, Kleman and Kim 12 ). Previous studies have reported that n-3 PUFA could beneficially affect lipid oxidation through phosphorylated AMPK (pAMPK) in the liver( Reference Suchankova, Tekle and Saha 13 ), adipose tissue and skeletal muscle( Reference Scorletti and Byrne 14 ). In addition, long-chain n-3 PUFA supplementation could activate AMPK and improve glucose uptake in the intestine( Reference Gabler, Radcliffe and Spencer 15 ). Nevertheless, these results could not lead to the conclusion that AMPK are indispensable for the effects of long-chain n-3 PUFA on FA oxidation. Moreover, no evidence has suggested any effects of n-3 PUFA on intestinal lipid metabolism through AMPK.
Another important point is that most of the previous studies were focused on DHA and EPA. However, the plant-derived α-linolenic acid (ALA) is more available than the marine-derived DHA and EPA, because of the cost and supply constraints of seafood( Reference Zhang, Li and Li 16 ). As a result, ALA is the most widely consumed n-3 PUFA in the Western diet( 17 ). It is also worth noting that fish oil (rich in DHA and EPA) may not completely reproduce the effects of ALA( Reference Finnegan, Minihane and Leigh-Firbank 18 ). Therefore, the present study was conducted to determine whether ALA could increase intestinal lipid metabolism and whether AMPK are necessary for these effects of ALA by using mice deficient in AMPKα1 and AMPKα2.
Methods
Animals
All mice were housed individually and maintained at 21±2°C, under a 12:12 h light–dark cycle with free access to water and food. To analyse the effects of ALA on intestinal lipid metabolism and to clarify whether AMPK was involved, we used AMPKα1–/– and AMPKα2–/– mice with a C57BL/6 genetic background. The AMPKα1 and AMPKα2 knockout mice were purchased from the Jackson laboratory and C57BL/6 mice were used as control.
Experimental design and sample collection
All mice at 8 weeks of age were maintained on a 45 % HF diet. The HF diet consisted of 45 % (kcal%) fat from lard and soyabean oil, 20 % protein and 35 % carbohydrate. FA composition of diet fat was presented in Table 1. C57BL/6, AMPKα1–/– and AMPKα2–/– mice (eight male mice per treatment) at 9 weeks of age were separately divided into two groups: either consuming a 45 % HF or 45 % HF containing 10 % ALA (Aladdin Ltd; maintaining the total amount of fat at 45%) for 12 weeks. Food intake was measured throughout the study every 3 d, and body weight was recorded every week. On the last day of the experiment, mice were killed by cervical dislocation and blood was collected from the eyeballs. Next, the small intestine was removed and the jejunum and ileum were separated, thoroughly fluxed with sterile saline and immediately frozen in liquid N2 and then stored at –80°C.
Table 1 Fatty acid (FA) composition of dietary fat

HF, high-fat diet; HF-A, high-fat diet supplemented with α-linolenic acid.
All experiments were approved by the Committee of Experimental Animal Care, Zhejiang University (Hangzhou, China).
Measurement of TAG and fatty acid composition in blood and faeces
TAG content was measured in dried faecal samples and presented as µg/d according to a previous study( Reference Kimura, Takahashi and Lin 7 ). Briefly, the faeces were dried at 60°C overnight and lipids were extracted by the method of Folch et al. ( Reference Folch, Lees and Sloane Stanley 19 ). Next, TAG content in the lipid extracted from faeces and in serum were assayed using a TAG assay kit (GPO-POD; Applygen Technologies Inc.) as Zhang et al. did( Reference Zhang, Yang and Guo 20 ). To analyse the FA composition of serum, lipid extraction and transesterification were processed according to a previous method( Reference Lepage and Roy 21 ). FA methyl esters were measured by GC. The FA were identified by comparing the retention time of standard esters, and the composition of FA was calculated as a percentage of the total area.
Quantitative RT-PCR analysis
Total RNA was extracted with the TRIzol reagent (Invitrogen) and then complementary DNA was synthesised with RevertAid Reverse Transcriptase (Fermentas). Real-time PCR was performed according to the method used previously( Reference Zhou, Chen and Wang 22 ). Briefly, the PCR system consisted of 10 µl of SYBR Premix Ex Taq (2×) mix (Roche), 0·4 µl of ROX (50×) (Roche), 1·0 µl of cDNA, 7·8 µl of double-distilled water and 0·4 µl of primer pairs (10 mm), all in a total volume of 20 µl. The PCR cycle involved an initial step at 95°C for 30 s, followed by 40 cycles of 5 s at 95°C and 34 s at 60°C. All primers were presented in Table 2. The comparative C t value method was used to quantify mRNA expression relative to 18s rRNA. All samples were run in triplicate, and the average values were calculated.
Table 2 Mouse primers used for RT-PCR analysis

CPT1a, carnitine palmitoyltransferase 1a; ACOX1, acyl-CoA oxidase 1; ACADM, medium-chain acyl-CoA dehydrogenase; Cyp4a10, cytochrome P450 4A10; pdk4, pyruvate dehydrogenase kinase isoenzyme 4; MGAT2, monoacylglycerol O-acyltransferase 2; DGAT1, diacylglycerol O-acyltransferase 1; DGAT2, diacylglycerol O-acyltransferase 2; CD36, cluster of differentiation 36; FATP4, fatty acid transport protein 4.
Western blotting analysis
Proteins were extracted and supernatants were resolved on 10 % SDS/PAGE gel, and then electro-blotted onto a polyvinylidene difluoride membrane. Primary antibodies against AMPKα1, AMPKα2, pAMPKα1 (Santa Cruz), pAMPKα2 (Abcam) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Boster) were applied overnight at 4oC. After incubating with the secondary antibody for 1 h at room temperature, the membrane was detected using the Enhanced Chemiluminescence Detection Kit for horseradish peroxidase (Biological Industries).
Statistical analysis
Data were analysed as a 3×2 factorial, except that RT-PCR data were analysed as a 3×2×2 factorial using Proc Mixed of SAS (SAS Institute Inc.). The statistical model for body weight gain, food intake and serum parameters include diet, genotype and their interaction. The statistical model for gene expression includes diet, genotype, intestinal segment and their interaction. Treatment means were calculated using the LSMEANS statement, and means are separated using the PDIFF option of PROC MIXED. An α-value of 0·05 was used to assess significant differences among means.
Results
Body weight gain and food intake in wild-type, AMPKα1–/– and AMPKα2–/– mice
According to Table 3, there was no effect of genotypes (wild type (WT), AMPKα1–/– and AMPKα2–/–) or type of diets (HF and HF diet supplemented with ALA (HF-A)) on food intake, and no interactions between genotypes and types of diet were observed. Moreover, no interactions between genotypes and types of diet were observed for body weight gain. When compared with mice fed an HF diet, mice fed an HF-A diet had less body weight gain (17.2±0.3 v. 13.2±1.1 g). However, there was no significant effect of genotypes on body weight gain.
Table 3 Body weight gain (BWG) and food intake (FI) in wild-type mice (WT), AMPKα1 whole-body knockout mice (AMPKα1–/–) and AMPKα2 whole-body knockout mice (AMPKα2–/–)
(Mean values with their pooled standard errors)

AMPK, AMP-activated protein kinase; HF, high-fat diet; HF-A, high-fat diet supplemented with α-linolenic acid; IBW, initial body weight.
TAG concentration in serum and faeces and serum fatty acid composition in wild-type, AMPKα1–/– and AMPKα2–/– mice
According to Table 4, there was a tendency for significant interactions between genotypes and types of diet for TAG concentration in serum. In fact, this tendency was reflected in WT mice only. In addition, there was no effect of genotypes or types of diet on faecal TAG output, and no interactions between genotypes and types of diet were observed. As no interactions between genotypes and types of diet were observed and the effect of the genotypes was never significant, the data of FA composition in serum were presented independently from the genotypes (Table 5). Compared with mice fed an HF diet, mice fed an HF-A diet had higher levels of ALA (C18 : 3), EPA (C20 : 5) and DHA (C22 : 6) in serum, whereas mice fed an HF-A diet had a lower level of C18 : 1.
Table 4 TAG content in serum and faeces of wild-type mice (WT), AMPKα1 whole-body knockout mice (AMPKα1–/–) and AMPKα2 whole-body knockout mice (AMPKα2–/–)
(Mean values with their pooled standard errors)
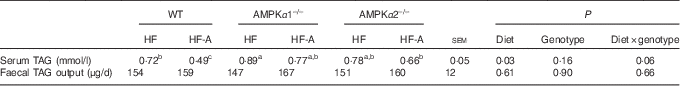
AMPK, AMP-activated protein kinase; HF, high-fat diet; HF-A, high-fat diet supplemented with α-linolenic acid.
a,b,c Within a row, means with unlike superscript letters tended to be significantly different.
Table 5 Fatty acid composition in serum of wild-type mice (WT), AMPKα1 whole-body knockout mice (AMPKα1–/–) and AMPKα2 whole-body knockout mice (AMPKα2–/–)Footnote *
(Mean values with their pooled standard errors)

AMPK, AMP-activated protein kinase.
HF: WT, AMPKα1–/– and AMPKα2–/– mice fed a high-fat diet; HF-A: WT, AMPKα1–/– and AMPKα2–/– mice fed a high-fat diet supplemented with α-linolenic acid.
a,b Mean values within a row with unlike superscript letters were significantly different (P<0·05).
* As no interactions between genotypes and types of diet were observed and the effect of the genotypes is never significant, the data are presented independently from the genotype.
Effects of α-linolenic acid on the protein expression of AMPK and pAMPK in the small intestine of mice
No interactions among genotypes, types of diet and intestine site and other interactions were observed for protein expression of AMPKα1 and AMPKα2 in both the jejunum and the ileum. Protein expression levels of AMPKα2 in AMPKα1–/– mice were higher than in WT mice, and protein expression levels of AMPKα1 in AMPKα2–/– mice were higher than in WT mice in both the jejunum and the ileum (Fig. 1(a)–(e)). No interactions among genotypes, types of diet and intestine site and other interactions were observed, except that interactions between genotypes and types of diet were observed for relative protein expression of pAMPKα1 and pAMPKα2 in both the jejunum and the ileum, because the HF-A diet increased relative protein expression of pAMPKα1 compared with the HF diet in WT mice but not in AMPKα2–/– mice, and HF-A diet increased relative protein expression of pAMPKα2 compared with the HF diet in WT mice but not in AMPKα1–/– mice (Fig. 1(f)–(j)).

Fig. 1. Protein expression of AMP-activated protein kinase (AMPK) and phosphorylated AMPK (pAMPK) in the jejunum and ileum of mice. For subparts (a), (b), (c), (d), (f), (g), (h) and (i), relative protein expression was analysed by ImageJ. In subparts (e) and (j), Western blot analysis. AMPKα1 KO, AMPKα1–/–, AMPKα1 whole-body knockout mice; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; WT, wild type; AMPKα2 KO, AMPKα2–/–, AMPKα2 whole-body knockout mice; HF, high-fat diet (![]() ); HF-A, high-fat diet supplemented with α-linolenic acid (
); HF-A, high-fat diet supplemented with α-linolenic acid (![]() ). a,b,c Mean values with unlike superscript letters were significantly different (P<0·05). No diet × genotype × site interactions and other interactions were detected (P>0·05) except that diet × genotype interactions were observed for relative protein expression of pAMPKα1 and pAMPKα2 in both the jejunum and ileum (P<0·05).
). a,b,c Mean values with unlike superscript letters were significantly different (P<0·05). No diet × genotype × site interactions and other interactions were detected (P>0·05) except that diet × genotype interactions were observed for relative protein expression of pAMPKα1 and pAMPKα2 in both the jejunum and ileum (P<0·05).
Effects of α-linolenic acid on gene expression of fatty acid oxidation, fatty acid transportation and TAG synthesis-related genes in the small intestine of mice
According to Table 6, no interactions among genotypes, types of diet and intestine site and other interactions were detected, except that interactions between genotypes and types of diet were observed for gene expression of CPT1a, acyl-CoA oxidase 1 (ACOX1), medium-chain acyl-CoA dehydrogenase (ACADM), cytochrome P450 4A10 (Cyp4a10), pyruvate dehydrogenase kinase isoenzyme 4 (pdk4), monoacylglycerol O-acyltransferase 2 (MGAT2), diacylglycerol O-acyltransferases 1 and 2 (DGAT1 and DGAT2) in both the jejunum and the ileum, because diet supplemented with ALA increased gene expression of CPT1a, ACOX1, ACADM (FA oxidation-related genes, which suggested an enhanced β-oxidation activity), Cyp4a10 (FA oxidation-related genes, which suggested an enhanced ω-oxidation activity), pdk4 (strongly suggested a switch from glycolysis to fatty acid oxidation)(23), MGAT2, DGAT1 and DGAT2 (TAG synthesis-related genes), but not in AMPKα2–/– and AMPKα1–/– mice. Gene expression levels of fatty acid transport protein 4 (FATP4) and the cluster of differentiation 36 (CD36) were higher in the jejunum than those in the ileum, whereas neither genotypes nor types of diet significantly affected gene expression of FATP4 and CD36.
Table 6 Relative gene expression of fatty acid (FA) oxidation, transportation and TAG synthesis-related genes in the jejunum and the ileum of mice with different genotypes and diet treatmentFootnote *
(Mean values with their pooled standard errors)

AMPKα1–/–, AMPKα1 whole-body knockout mice; AMPKα2–/–, AMPKα2 whole-body knockout mice; HF, high-fat diet; HF-A, high-fat diet supplemented with α-linolenic acid.
CPT1a (carnitine palmitoyltransferase 1a), ACOX1 (acyl-CoA oxidase 1) and ACADM (medium-chain acyl-CoA dehydrogenase) are involved in β-oxidation activity; Cyp4a10 (cytochrome P450 4A10) is involved in ω-oxidation activity; high expression of pdk4 (pyruvate dehydrogenase kinase isoenzyme 4) suggests a switch from glycolysis to FA oxidation; CD36 (the cluster of differentiation 36) and FATP4 (fatty acid transport protein 4) are involved in fatty acid transportation; MGAT2 (monoacylglycerol O-acyltransferase 2), DGAT1 and DGAT2 (diacylglycerol O-acyltransferase 1 and 2) are involved in TAG synthesis.
a,b Mean values within a row with unlike superscript letters were significantly different (P<0·05).
* No diet×genotype×site interactions and other interactions were detected (P>0·05), except that diet×genotype interactions were observed for gene expression of CPT1a, ACOX1, ACADM, Cyp4a10, pdk4a, MGAT2, DGAT1 and DGAT2 in both the jejunum and ileum (P<0·05).
Discussion
n-3 Long-chain PUFA, DHA and EPA have been demonstrated to increase intestinal lipid metabolism and attenuate hyperlipidaemia( Reference van Schothorst, Flachs and Franssen-van Hal 5 – Reference Kimura, Takahashi and Lin 7 ). However, little research concern the effects of their precursor ALA on intestinal lipid oxidation, except that ALA-rich TAG could reduce body weight gain and stimulate the intestinal β-oxidation( Reference Murase, Nagasawa and Suzuki 24 ). In the current study, our results showed that ALA alone could protect WT mice from HF-induced hyperlipidaemia and increased intestinal FA oxidation.
Some studies demonstrate that the efficiency of ALA conversion to DHA and EPA is very limited( Reference Jafari, Fallah and Azadbakht 25 ), whereas other studies report that dietary ALA could increase the levels of EPA and DHA in serum, adipose tissue( Reference Gonzalez-Manan, Tapia and Gormaz 26 ) and hepatic membrane( Reference Hanke, Zahradka and Mohankumar 27 , Reference Kim and Choi 28 ). Moreover, ALA, without converting to DHA and EPA, could protect against HF-induced obesity and its related diseases( Reference Monteiro, Askarian and Nakamura 29 ). Our results showed that dietary ALA decreased HF-induced serum TAG increase in WT mice fed an HF diet. Dietary ALA remarkably improved serum ALA level in either WT mice or AMPK-lacking mice. In addition, a low but measurable level of DHA and EPA were observed in serum after ALA supplementation. Consequently, our results further confirmed that ALA alone could alleviate hyperlipidaemia, as previous study demonstrated( Reference Monteiro, Askarian and Nakamura 29 ). In addition, genotypes had no effects on FA composition in serum.
AMPK is a heterotrimeric enzyme consisting of an α catalytic subunit and noncatalytic β and γ subunits. The α1 and α2 isoforms of the α subunit share 90 % amino acid sequence homology within the catalytic site, but major differences in the C-terminal tails of α1 and α2 sequences exist( Reference Stapleton, Mitchelhill and Gao 30 ). As a result, these two catalytic subunits conferred tissue-specific difference in regard to the formation of heterotrimers and metabolic regulation( Reference Ceddia 31 ). For the recent decades, some findings suggested that the α2 subunit was responsible for the modulation of gene expression( Reference Salt, Johnson and Ashcroft 32 ). However, other results indicated that the α1 catalytic subunit accounts for most of the activity of AMPK, especially in adipocytes( Reference Daval, Diot-Dupuy and Bazin 33 , Reference Lihn, Jessen and Pedersen 34 ). Moreover, mice lacking the α2 subunit exhibited adiposity and adipocyte hypertrophy when subjected to the HF diet( Reference Villena, Viollet and Andreelli 35 ). Our results showed that when one catalytic subunit was lacking, the expression of the other catalytic subunit was compensatorily up-regulated. Surprisingly, ALA did not affect the expression of AMPKα subunit in the intestine. However, ALA supplementation increased AMPK phosphorylation of both α1 and α2 subunit in the small intestine. These results suggested that ALA may increase intestinal AMPK activity.
Furthermore, mice with a whole-body deletion of AMPKα2–/– and feeding on HF had the same serum TAG level as WT mice, as previously described( Reference Villena, Viollet and Andreelli 35 ). In our results, the observation that AMPKα2–/– mice also had the same expression of genes involved in FA oxidation in the intestine as WT mice when fed an HF diet indicated a compensatory effect in the AMPK catalytic subunit. However, when subjected to an HF-A, WT mice tended to have a lower level of serum TAG than AMPKα2–/– mice and mRNA expression levels of genes involved in FA oxidation in the intestine of WT mice was higher than those in AMPKα2–/– mice. These results indicated that AMPKα2 was required for the effects of ALA on intestinal FA metabolism. It has been reported that mice lacking AMPKα2–/– showed an enhanced lipid accumulation in adipocytes without any changes in adipocytes number or differentiation( Reference Villena, Viollet and Andreelli 35 ). However, both our results and those from the previous study observed no significant difference of serum TAG concentration between WT and AMPKα2–/– mice fed an HF. Accordingly, one of the important reasons for the low serum TAG level of WT mice subjected to an HF-A may therefore be the increased FA oxidation in the small intestine, although increased FA oxidation in small intestine would not be the only reason as ALA could also increase lipid oxidation in other tissues such as adipose tissue( Reference Fukumitsu, Villareal and Onaga 36 ), liver( Reference Gonzalez-Manan, Tapia and Gormaz 26 ) and skeletal muscle( Reference Oliva, Ferreira and Chicco 37 ). In contrast to the previous report, demonstrating that AMPKα2 is not necessary for hypolipidaemic effect of n-3 PUFA (DHA and EPA) in ad libitum-fed mice( Reference Jelenik, Rossmeisl and Kuda 38 ), our study showed a tendency of higher level of serum TAG in AMPKα2–/– mice fed an HF-A than that in WT mice. These results indicated that AMPKα2 may only be involved in the effects of ALA, but not in the effects of DHA or EPA. However, mice with a whole-body deletion of AMPKα1–/– and feeding on HF tended to have a higher TAG level in serum than WT mice, and ALA addition in the diet did not decrease TAG level in serum in AMPKα1–/– mice. These results indicated that AMPKα1 was required for protection by ALA from HF-induced hyperlipidaemia. Moreover, the fact that ALA supplementation increased mRNA expression of genes involved in FA oxidation only in WT mice but not in AMPKα1–/– mice indicated that AMPKα1 is also indispensable for the effects of ALA on intestinal FA oxidation.
In addition, neither ALA addition nor AMPK catalytic subunit deletion affected the expression of genes involved in intestinal FA uptake, as no significant changes were observed in CD36 and FATP4 expression. Moreover, no difference in faecal TAG output in all treatments could be further evidenced. However, TAG synthesis in WT mice subjected to HF-A diet was significantly decreased, as mRNA expression of DGAT1 and DGAT2 was higher than with the other treatment. In addition, increased FA oxidation caused by ALA supplementation may be the primary reason for this change.
In conclusion, our results showed that ALA protected WT mice from HF-induced hyperlipidaemia and increased intestinal FA oxidation. Moreover, both AMPKα1 and AMPKα2 were indispensable for the effects of ALA on intestinal FA oxidation. These findings might provide new sights to better understand the effects of ALA on intestinal FA metabolism and their molecular mechanisms, and would be useful in the management of systemic lipid metabolism. Nevertheless, further studies will be needed to explore the differential effects of n-6 PUFA and n-3 PUFA on intestinal lipid metabolism and their molecular mechanisms.
Acknowledgements
This work was financially supported by National Basic Research Program of China (grant no. 2012CB124705).
X. Z. and J. C. designed and carried out the experiment, analysed the data and wrote the manuscript. W. W. carried out the experiment. X. W. contributed to the experimental design and discussion. Y. W. contributed to the experimental design, discussion and manuscript modification.
None of the authors has either financial or personal conflicts of interest to declare.







