


Horses have evolved from ancestors with an intestinal system designed to process large quantities of low-quality forage, containing high levels of structural plant polysaccharides, ingested on an almost continuous basis to meet their nutrient demands. The large intestine (hindgut) of the horse is anatomically specialised to accommodate micro-organisms capable of degrading and fermenting structural polysaccharides of the plant cell wall, which are generally resistant to pre-caecal digestion(Reference Hainze, Muntifering and McCall1). The fermentation of feedstuffs in the hindgut results in the production of volatile fatty acids that, when absorbed, constitute a significant proportion (30 %) of the digestible energy intake of the animal; particularly in horses fed high-fibre diets(Reference Glinsky, Smith, Spires and Davis2). Fibre-based diets are known to maintain normal fermentation conditions within the large intestine whilst, in contrast, diets containing high levels of concentrates (starch) can be detrimental to the maintenance of a homeostatic hindgut environment(Reference McLean, Hyslop, Longland, Cuddeford and Hollands3), and can lead to a number of metabolic disorders such as acidosis and laminitis(Reference Carroll, Hazard, Coloe and Hooper4–Reference Rowe, Lees and Pethick7).
Despite the importance of the intestinal microbial ecosystem in many aspects of host animal health and performance in other species, particularly ruminants, there is a dearth of information regarding the microbial ecology of the equine hindgut. A greater understanding of the microbial diversity of the hindgut is essential for improving our knowledge of digestive processes, and for the future prevention and treatment of diseases involving the gastrointestinal tract, for example, laminitis and grass sickness. However, knowledge of the bacterial populations present in the large intestine of the horse is very limited, compared, for example, with the rumen(Reference Tajima, Aminov, Nagamine, Ogata, Nakamura, Matsui and Benno8) and the caecum and colon of humans and pigs(Reference Wilson and Blitchington9, Reference Pryde, Richardson, Stewart and Flint10). Current knowledge of gut microbial ecology and diversity is almost exclusively based on the use of classic culture-based methods that are often laborious, time consuming and may only recover a fraction of the microbial diversity present within the gut(Reference Daly, Stewart, Flint, Soraya and Shirazi-Beechey11). However, advanced modern molecular methods, such as real-time semi-quantitative PCR (Q-PCR), are culture-independent tools for accurate and sensitive quantification of individual bacterial species as well as total bacterial numbers(Reference Halliwell, Fleischman, Mackay-Smith, Beech and Gunson12, Reference Nadkarni, Martin, Jacques and Hunter13). Limited studies have reported the bacterial diversity within the large intestine of the horse using more conventional molecular methods such as end-point PCR or use of oligonucleotides(Reference Daly, Stewart, Flint, Soraya and Shirazi-Beechey11, Reference Julliand, de Vaux, Millet and Fonty14, Reference Daly, Soraya and Shirazi-Beechey15), with these studies identifying the fibrolytic bacteria Ruminococcus flavefaciens and Fibrobacter succinogenes predominating. In these early hybridisation studies investigating the equine hindgut, some authors have lyophilised the material in order to account for the DM increase along the intestinal tract(Reference Julliand, de Vaux, Millet and Fonty14), whilst others have extracted DNA from frozen material(Reference Daly, Stewart, Flint, Soraya and Shirazi-Beechey11, Reference Daly, Soraya and Shirazi-Beechey15). However, it is unclear if the preservation method affects the data obtained from studies using PCR methodologies and whether this should be considered when interpreting results from frozen and lyophilised material.
Furthermore, to our knowledge, there are no published data on the identification or quantification of equine intestinal bacteria using real-time PCR technology, which is a more accurate and sensitive alternative to conventional end-point PCR-based methodologies, and has recently been applied to study diet-dependent shifts in the bacterial populations of the rumen(Reference Tajima, Aminov, Nagamine, Matsui, Nakamura and Benno16) and infant gut(Reference Haarman and Knol17). Moreover, previous studies investigating microbial diversity and fermentation characteristics within the equine hindgut typically used animals specifically euthanased for the purpose, or surgically modified animals. Whilst these methods have provided important insights into the equine microbial ecosystem, they can be expensive and there is a consensus nowadays to adopt, where possible, cost-effective welfare-friendly alternatives.
Consequently, the objectives of the work reported here were to: (1) optimise real-time Q-PCR methodologies for quantifying changes in relative amounts of R. flavefaciens, F. succinogenes (fibrolytic bacteria) and Streptococcus bovis (non-fibrolytic bacterium) in the luminal contents of the equine caecum, ventral colon, dorsal colon and rectum; (2) compare the relative amounts of these candidate bacteria in frozen and lyophilised samples; (3) establish whether faeces are a suitable model of hindgut function in the horse. The candidate bacteria are likely to play key roles in equine digestion and health, given that F. succinogenes and R. flavefaciens are key fibrolytic bacteria, whilst the saccharolytic bacterium S. bovis has been proposed as having a role in hindgut acidosis and laminitis(Reference Rowe, Lees and Pethick7, Reference Julliand, de Vaux, Millet and Fonty14, Reference Milinovich, Trott, Burrell, Thoefner, Blackall, Al Jassim, Morton and Pollitt18).
Materials and methods
Collection and processing of samples
Samples of luminal contents (caecum; ventral colon; dorsal colon; rectum) were taken from fourteen freshly slaughtered horses (unknown age and breed), not suffering from any known intestinal diseases, obtained from the local abattoir. Upon recovery, lumen contents were placed in individually labelled grip-top bags and immediately placed on dry ice. At the laboratory, each sample was sub-divided into two groups; one of which remained frozen and was stored at − 80°C until required, whilst the other was lyophilised to constant weight before storage at − 80°C. Quantification was carried out in luminal contents only, as previous work has established that microbial community structure between the hindgut wall and lumen contents is not different in equines(Reference Daly, Stewart, Flint, Soraya and Shirazi-Beechey11).
DNA extraction
Total DNA extraction from frozen and lyophilised luminal samples was carried out using the QIAamp® DNA stool kit (Qiagen Ltd, Crawley, West Sussex, UK). This purifies genomic, bacterial, viral and parasite DNA from stool samples and was used in accordance with the manufacturer's recommendations, with some modifications. The following procedure was carried out for each sample of frozen and lyophilised lumen contents.
Lumen contents (180–220 mg) were homogenised in 1·4 ml of buffer ASL using a RiboLyser (Hybaid Ltd, Ashford, Middlesex, UK). Following homogenisation each sample was heated at 95°C for 5 min to lyse the bacteria, and then centrifuged for 3 min at 14 000 g. The supernatant fraction was removed and placed into a microcentrifuge tube where it was vortexed with an InhibitEX tablet for 1 min, or until the tablet was completely suspended. The suspension was then incubated for 1 min at room temperature to allow potential PCR inhibitors or DNA-degrading substances to absorb to the InhibitEX matrix. The InhibitEX reagent was then pelleted by centrifugation for 3 min at 14 000 g, after which 200 μl of the supernatant fraction was then removed and placed in a new microcentrifuge tube containing 15 μl proteinase K. Then 200 μl buffer AL was added and the mixture was thoroughly vortexed for 15 s. After further heating at 70°C for 10 min to allow protein digestion and degradation under denaturing conditions, 200 μl absolute ethanol was added. The resultant mixture was then loaded onto a QIAamp® spin column (Qiagen Ltd) and centrifuged for 3 min at 14 000 g. The DNA bound to the spin column was then washed in two centrifugation steps, first with 500 μl of buffer AW1 followed by 500 μl buffer AW2, at 14 000 g for 1 min and 3 min, respectively. Finally, purified DNA was eluted from the spin column in 200 μl of buffer AE by allowing it to incubate for 1 min at room temperature, followed by centrifugation at 14 000 g for 1 min. DNA was stored at − 20°C until required for real-time PCR.
Real-time polymerase chain reaction
Semi-quantitative real-time PCR was performed on extracted DNA from the frozen and lyophilised luminal contents for R. flavefaciens, F succinogenes, S. bovis and total bacterial load, using the MX3000P Q-PCR system (Stratagene Ltd, Cambridge, Cambs, UK). The PCR reaction contained 10 × Thermo-Start® standard buffer, 25 mm-MgCl2, 5 mm each dNTP, Thermo-Start® DNA polymerase (Abgene Ltd, Epsim, Surrey, UK), 300 mm each forward and reverse primer, 200 mm probe, DNA template (from frozen or lyophilised contents) and molecular biological-grade water (BDH, Poole, Dorset, UK). Thermal cycling conditions were 2 min at 50°C followed by 10 min at 95°C and forty cycles of 15 s at 95°C and 2 min at 60°C. Samples were run in duplicate for each quantification assay.
Taqman® probes and oligonucleotide primers for R. flavefaciens, F. succinogenes and S. bovis were designed using Primer Express® software (PE Applied Biosystems, Warrington, Ches, UK). Probe and primer sets were designed based on R. flavefaciens, F. succinogenes and S. bovis 16S rDNA sequences published in GenBank®. Probes and primers were tested for specificity using the Basic Local Alignment Search Tool (BLAST; National Center for Biotechnology Information (NCBI), Bethesda, MD, USA). A previously published universal primer and probe set was used for the determination of the total bacterial load(Reference Nadkarni, Martin, Jacques and Hunter13). The probes and primers were synthesised by MWG-Biotech AG (Ebersberg, Germany). All probes contained 6-carboxy-fluorescein as the 5′ reporter and 6-carboxy-tetramethyl-rhodamine (TAMRA) as the 3′ quencher. Details of primers and probes are given in Table 1.
Table 1 Bacterial strains and GenBank® accession numbers utilised, with oligonucleotide PCR primers and Taqman® probe sequences used during real-time PCR, including amplicon length generated

For relative quantification of R. flavefaciens, F. succinogenes and S. bovis the comparative cycle threshold (CT; Fig. 1) method was used(19), which involved normalisation of the number of target copies to total bacterial load (universal). The ΔCT was first calculated (universal mean CT – specific bacteria mean CT). The normalised level of abundance was calculated using the formula = 1·78− ΔCT, where 1·78 was derived from Nadkarni et al. (Reference Nadkarni, Martin, Jacques and Hunter13) as the response to standard amounts of DNA obtained with the universal primer and probe set. As the target bacteria were generally represented in small proportions relative to total bacterial load, data were transformed by multiplying by 1000 to allow for ease of data handling. Validation studies were carried out to demonstrate that the amplification efficiencies of the universal primers and probe set and specific bacteria were equivalent. This involved generating relative standard curves for each primer and probe set using serial dilutions of purified DNA. The ΔCT (y) between the universal set and each specific bacterium was plotted v. log (dilution; x) to calculate the slope of the line (by linear regression analyses). Slopes for R. flavefaciens, F. succinogenes and S. bovis were all < 0·1 as required (Fig. 2).

Fig. 1 A typical example of amplification plots obtained during the real-time PCR reaction for the universal (–●–), Ruminococcus flavefaciens (–■–), Fibrobacter succinogenes (–▲–) and Streptococcus bovis (–♦–) primer and probe sets. The horizontal line represents the threshold fluorescence and corresponds to the cycle threshold value for a given sample. dR, baseline-subtracted fluorescence.
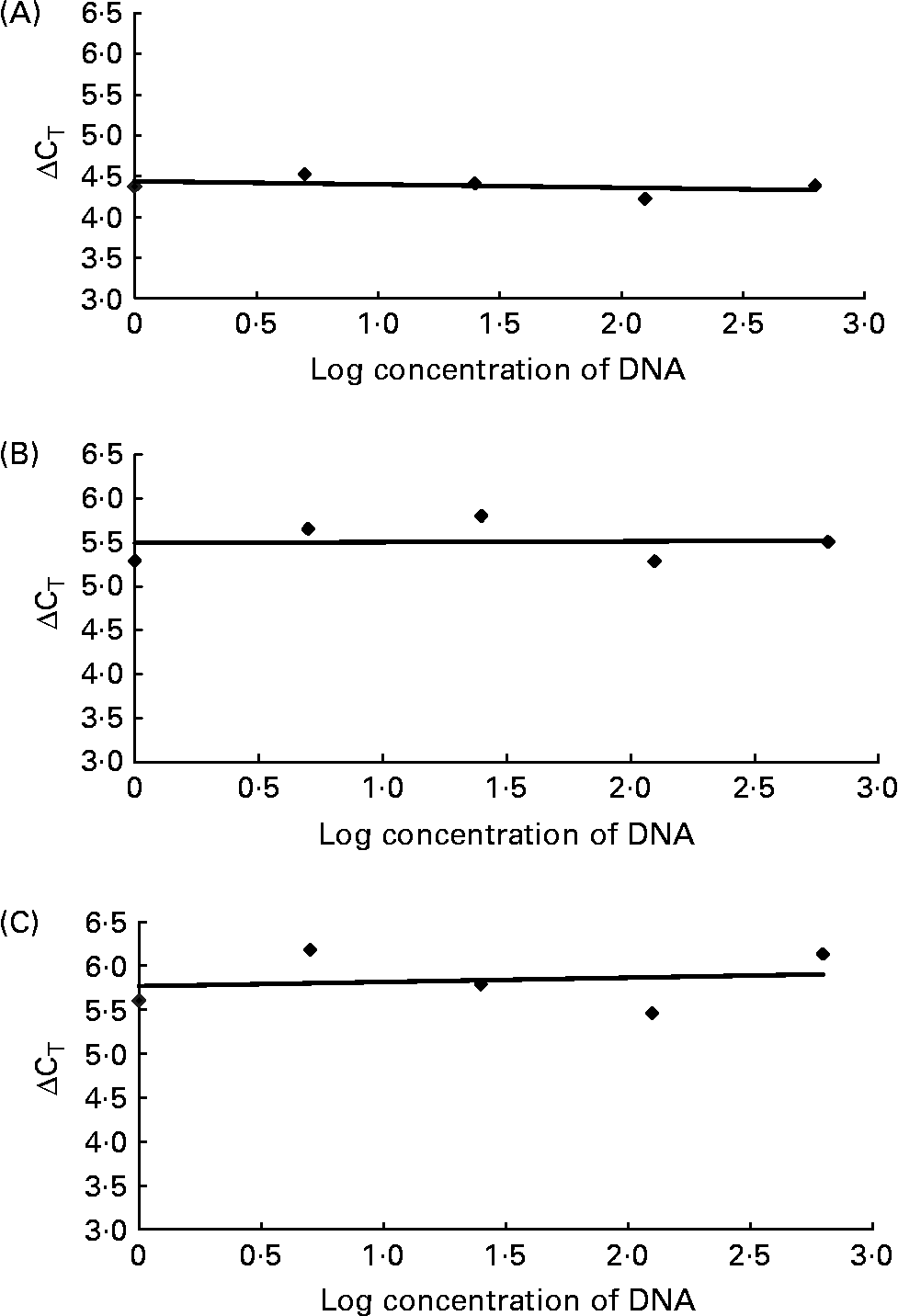
Fig. 2 Validation studies demonstrating the amplification efficiencies of the universal primer and probe set to those of Ruminococcus flavefaciens (A; y = − 0·0401x), Fibrobacter succinogenes (B; y = 0·0093x) and Streptococcus bovis (C; y = 0·0486x). The slope of each line was calculated by linear regression analysis. The absolute value of the slope was close to zero (y = < 0·1), therefore the efficiencies of the target and reference genes were similar. CT, cycle threshold.
Statistical analyses
Data generated from real-time PCR were not normally distributed; a logarithmic (base 10) transformation was therefore performed on all of the data before statistical analysis. Values for the relative amounts of bacteria in the various regions of the hindgut were analysed for significant differences using two-way ANOVA in GenStat® release 9.1 (Lawes Agricultural Trust, Harpenden, Herts, UK). This was done separately for both the frozen and lyophilised material. Values for bacterial species and preservation treatment (frozen or lyophilised) were also analysed for significant differences using two-way ANOVA. Comparisons between treatment groups were made by least significant difference equations. P values of < 0·05 were considered statistically significant.
Results
The quantification of R. flavefaciens, F. succinogenes and S. bovis, involving the normalisation of the number of target copies to total bacterial load, confirmed the application of real-time PCR to successfully detect R. flavefaciens, F. succinogenes and S. bovis from the equine hindgut (Fig. 1). The relative quantification of R. flavefaciens, F. succinogenes and S. bovis 16S rDNA extracted from the large intestine of fourteen healthy horses confirmed that these bacterial species are all abundant at detectable levels throughout the equine hindgut, differing in relative quantification from region to region.
Analysis of real-time data showed no significant interaction between bacterial species and hindgut region; therefore, main effects were examined in isolation. Data derived from real-time PCR revealed that region of the equine hindgut significantly affected the overall bacterial load of R. flavefaciens, F. succinogenes and S. bovis in both frozen (P = 0·011; Table 2) and lyophilised (P < 0·001; Table 3) luminal contents. Overall, caecal samples had significantly (P < 0·01) fewer R. flavefaciens, F. succinogenes and S. bovis than were present in the luminal contents of the ventral colon, dorsal colon and rectum in both the frozen and lyophilised samples. However, similar candidate bacterial loads were observed between the luminal contents of ventral colon, dorsal colon and rectum in the frozen samples (Table 2). In contrast, in the lyophilised samples significantly fewer R. flavefaciens, F. succinogenes and S. bovis were present in the luminal contents of the ventral colon (P < 0·05; Table 3), compared with the dorsal colon and rectum, which were similar.
Table 2 Semi-quantitative levels of Ruminococcus flavefaciens, Fibrobacter succinogenes and Streptococcus bovis in frozen luminal contents of the equine caecum, ventral colon, dorsal colon and rectum (n 14)*
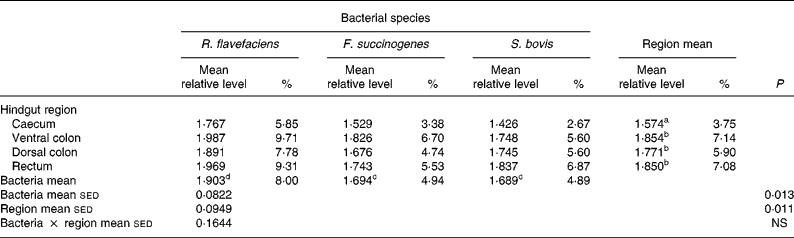
sed, Standard error of the difference, CT, cycle threshold.
a,b Mean values within a column with unlike superscript letters were significantly different (P < 0·05).
c,d Mean values within a row with unlike superscript letters were significantly different (P < 0·05).
* Data (log-transformed) are expressed relative to mean total bacterial load. Actual percentages of total bacterial load are also shown. Log-transformed data were derived according to the formula = log10(1000(1·78− ΔCT)).
Table 3 Semi-quantitative levels of Ruminococcus flavefaciens, Fibrobacter succinogenes and Streptococcus bovis in lyophilised luminal contents of the equine caecum, ventral colon, dorsal colon and rectum (n 14)*
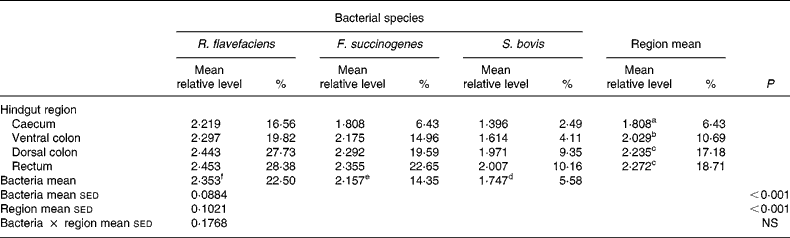
sed, Standard error of the difference, CT, cycle threshold.
a,b,c Mean values within a column with unlike superscript letters were significantly different (P < 0·05).
d,e,f Mean values within a row with unlike superscript letters were significantly different (P < 0·05).
* Data (log-transformed) are expressed relative to mean total bacterial load. Actual percentages of total bacterial load are also shown. Log-transformed data were derived according to the formula = log10(1000(1·78− ΔCT)).
With respect to the three individual bacteria, R. flavefaciens was the predominant bacterial species within each region of the equine hindgut sampled, for both the frozen and lyophilised material, and overall was present in significantly greater amounts than both F. succinogenes (P < 0·05) and S. bovis (P < 0·01) (Tables 2 and 3). However, in the frozen samples values for F. succinogenes and S. bovis were similar (Table 2), whilst in the lyophilised samples F. succinogenes was present at significantly (P < 0·05) higher levels than S. bovis throughout each region of the hindgut (Table 3).
Further analysis of real-time data revealed a significant (P < 0·001) interaction between bacterial species and preservation treatment (Table 4). Values for R. flavefaciens and F. succinogenes were significantly (P < 0·05) higher in the lyophilised material compared with the frozen samples, whereas similar values were obtained for S. bovis in both the frozen and lyophilised material.
Table 4 Semi-quantitative levels of Ruminococcus flavefaciens, Fibrobacter succinogenes and Streptococcus bovis in lyophilised and frozen luminal contents of the equine hindgut (n 14)*
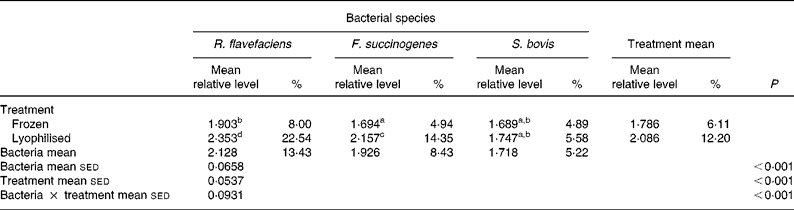
sed, Standard error of the difference, CT, cycle threshold.
a,b,c,d Mean values within a row or column with unlike superscript letters were significantly different (P < 0·05).
* Data (log-transformed) are expressed relative to mean total bacterial load. Actual percentages of total bacterial load are also shown. Log-transformed data were derived according to the formula = log10(1000(1·78− ΔCT)).
Discussion
The successful quantification of specific candidate bacteria involving the normalisation of the number of target copies to total bacterial load confirmed the efficacy of real-time PCR to successfully detect these bacteria within the equine hindgut. Semi-quantitative analysis of R. flavefaciens, F. succinogenes and S. bovis required specific probe and primer sets that were designed to target 16S rDNA using published sequences from bacterial strains that had already been identified in the equine(Reference Julliand, de Vaux, Millet and Fonty14, Reference Whitehead and Cotta20). In addition to these specific bacterial sets, a universal probe and primer set was also utilised that had a broad inter-species specificity capable of detecting as many of the bacterial populations within the equine hindgut as possible. However, modern molecular techniques such as real-time PCR have been used in only a limited number of studies, with few papers focusing on the whole equine bacterial community(Reference Daly, Stewart, Flint, Soraya and Shirazi-Beechey11, Reference Daly, Soraya and Shirazi-Beechey15); consequently, there are no equine-specific universal primer and probe sets. Therefore, the universal set used for the present study was a previously published set that had been designed to specifically detect the major groups of bacteria, as listed by Bergey's Manual of Determinative Bacteriology (Reference Nadkarni, Martin, Jacques and Hunter13). It is possible, however, that certain groups of equine-specific bacteria may not have been detected and/or efficiently amplified by this universal set. A high degree of genetic diversity has been reported for the bacterial community present in the equine hindgut(Reference Daly, Stewart, Flint, Soraya and Shirazi-Beechey11, Reference Milinovich, Trott, Burrell, Thoefner, Blackall, Al Jassim, Morton and Pollitt18), with novel clusters identified; these included clusters that were only represented by equine sequences and as such may be regarded as equine-only groups. However, there is also a great deal of microbiological diversity within the gastrointestinal tract in other species, and previous studies have already established the use of the universal primer and probe set to monitor bacterial load in gut contents(Reference Haarman and Knol17). Further molecular research using several primer sets would be required for an exhaustive survey of the microbial diversity from which an equine-specific universal probe and primer set could be designed. Nevertheless, the data reported here are semi-quantitative and for the first time compare R. flavefaciens, F. succinogenes and S. bovis relative to the total bacterial load throughout the various regions of the equine hindgut.
From the early hybridisation studies investigating the equine hindgut, some authors(Reference Julliand, de Vaux, Millet and Fonty14) have lyophilised the material in order to account for the DM increase along the intestinal tract. However, there is no information available in the literature on the effect of preservation method on data obtained from studies using PCR methodologies. One of the aims within the present study was to investigate the possible implications of lyophilising material before extracting the DNA. A very important finding in the present study was that significantly higher values were obtained for R. flavefaciens and F. succinogenes in the lyophilised material compared with the non-lyophilised samples, with no differences detected between preservation methods for S. bovis. It is unclear why these differences occurred; however, they may be attributed to differences in the liquid- and solid-associated bacteria. For instance, higher fibrolytic activities have been reported in the solid-associated bacteria in the equine hindgut(Reference Michalet-Doreau, Fernandez, Peyron and Millet21). In addition, freezing per se is unlikely to explain the differences observed, as the freezing occurs in both methods of preservation. Rather, it is possible that differences in preservation method may also be attributable to enzymic degradation of DNA, which may be released from lysed cells during subsequent thawing of frozen material before sub-sampling. Consequently, the preservation method of samples appears to be an important consideration in the enumeration of bacteria using this methodology as this could potentially have implications for quantifying and comparing results obtained within and across studies. It is also important to note that data from frozen samples in the present study were comparable with previous reports in the literature(Reference Julliand, de Vaux, Millet and Fonty14), whilst the abundance of the three candidate bacteria determined from lyophilised material were markedly higher than previous reports.
Real-time PCR data revealed frozen caecal contents had lower levels of the three candidate bacteria compared with the contents obtained from the ventral colon, dorsal colon and rectum. This is comparable with earlier observations established in culture-based experiments whereby a lower concentration of total anaerobic bacteria has been detected in the caecum(Reference de Fombelle, Julliand, Drogoul and Jacotot22). Previous molecular analysis of caecal contents suggests R. flavefaciens to be the predominant cellulolytic bacterial species in the equine caecum, with lower abundance reported for F. succinogenes (Reference Julliand, de Vaux, Millet and Fonty14). This concurs with the findings of the present study whereby the relative abundance of R. flavefaciens was notably higher than F. succinogenes in all regions of the large intestine in both the frozen and lyophilised material. However, the relative abundance reported for R. flavefaciens in the frozen caecal samples in the present study (5·85 %) is lower than the 9 % reported by Julliand et al. (Reference Julliand, de Vaux, Millet and Fonty14). Daly also detected R. flavefaciens in the various regions of the equine hindgut, but did not recover any sequences relating to F. succinogenes (Reference Daly, Stewart, Flint, Soraya and Shirazi-Beechey11), whilst Lin & Stahl(Reference Lin and Stahl23) concluded that F. succinogenes accounted for 12 % of total rRNA extracted from the caecum, which contrasts with the 3·38 and 6·43 % reported for frozen and lyophilised caecal samples, respectively, in the present study. Nevertheless, differing methodologies employed in the various studies may explain these conflicting results as well as differences in the host animal diet. Interestingly, DNA extracted from F. succinogenes has been shown to amplify less efficiently than other gut bacteria(Reference Tajima, Aminov, Nagamine, Matsui, Nakamura and Benno16); furthermore, diet has also been shown to play a major role in the biodiversity of microbial populations in the equine hindgut, with F. succinogenes appearing to thrive on low-quality roughage, unlike that of many other cellulolytic bacteria(Reference Koike, Shingu, Inaba, Kawai, Kobayashi, Hata, Tanaka and Okubo24). Moreover, the levels reported by Lin & Stahl(Reference Lin and Stahl23) were from a single animal, while Julliand et al. (Reference Julliand, de Vaux, Millet and Fonty14) reported a marked inter-animal variation in the percentage of F. succinogenes present in the caecum of horses fed identical diets. However, a limitation of the present study was that the nutritional history of the horses was unavailable.
One of the novel findings presented in the present study was the determination of the main lactic acid bacteria, S. bovis. To the best of our knowledge the abundance of this bacteria has yet to be determined within the equine hindgut, although several cultivation studies have focused on the lactobacilli and streptococci bacterial groups as a whole(Reference de Fombelle, Julliand, Drogoul and Jacotot22, Reference de Fombelle, Varloud, Goachet, Jacotot, Philippeau, Drogoul and Julliand25). Real-time PCR data revealed S. bovis to be present in lower amounts in the caecum compared with the ventral colon, dorsal colon and rectum in both the frozen and lyophilised material. This concurs with the findings of culture-based experiments, whereby, on average, the concentration of lactobacilli and streptococci tends to be lower in the caecum than the colon, which has been attributed to the faster rate of passage of soluble carbohydrate and undigested starch through the caecum compared with the colon(Reference de Fombelle, Julliand, Drogoul and Jacotot22).
Although S. bovis is a normal inhabitant of the gastrointestinal tract of the horse(Reference Al Jassim and Rowe26) it has been implicated as a putative causative agent for equine hindgut acidosis and related conditions, such as laminitis(Reference Milinovich, Trott, Burrell, Thoefner, Blackall, Al Jassim, Morton and Pollitt18). These conditions often arise as a result of dietary changes that lead to the proliferation of S. bovis and the production of excessive levels of lactic acid in the hindgut. S. bovis ferments non-structural carbohydrates and produces lactate as its main fermentation endproduct during rapid multiplication, eventually leading to decreases in hindgut pH. Consequently, diets high in non-structural carbohydrates can be detrimental to the maintenance of a homeostatic hindgut environment(Reference McLean, Hyslop, Longland, Cuddeford and Hollands3), and in severe cases this can lead to the death of the animal. The present paper has highlighted the potential uses of modern molecular technologies in exploring the role of S. bovis in equine gastrointestinal disease. Since 1952, S. bovis has been studied more extensively than any other lactic acid-producing bacteria of ruminal origin, with numerous strains having been isolated from cattle and sheep, characterised on both morphological and biochemical characteristics(Reference Russell and Robinson27, Reference Cotta28). However, various strains have been identified that do not cause disease(Reference Whitehead and Cotta20). As such, cultivation methods cannot be used as a reliable way to detect rising levels of S. bovis as the cause of gastrointestinal disease. More recently, S. bovis has been characterised on a molecular level in other species, using similar techniques to those presented here(Reference Whitehead and Cotta29, Reference Al Jassim, Scott, Trebbin, Trott and Pollitt30). However, to date this is the only study that has determined the abundance of S. bovis in equine hindgut contents. Nevertheless, further studies are required in equines to investigate the role that S. bovis plays in fermentative acidosis, gastric ulceration and laminitis. Furthermore, studies also need to consider other lactate-producing bacteria, since S. bovis is unlikely to be the sole bacterial species involved in gastrointestinal disease.
By establishing a model of hindgut function using non-invasive techniques, further research can explore the role of S. bovis, and other key bacteria, in different stages of gastrointestinal disease, and not just at the terminal stages following euthanasia. Data from both the frozen and lyophilised samples showed similar levels of R. flavefaciens, F. succinogenes and S. bovis relative to total bacterial load in luminal contents obtained from the dorsal colon and rectum. These findings indicate that, similar to other single-stomached animals(Reference Whitehead and Cotta20), equine faecal material could reflect the microbiological characteristics of the distal colon. This would subsequently allow faeces to act as a model for the distal colon, facilitating accurate determination of changes in gut microflora without the need for surgically modified animals or the use of slaughter material, which allows for no information on the animal's health or dietary management. Furthermore, data from the frozen luminal contents indicated similarities between the three bacteria in the ventral colon, dorsal colon and rectum, potentially allowing faeces to be used as a model for the whole colon. However, more work is required to further develop real-time Q-PCR for quantification of a greater number of candidate bacterial species, in particular key fibrolytic species and lactate-producing bacteria (especially from the genus Streptococcus), and to assess the effect of environmental factors, such as diet, on the relationship between faecal and colonic bacterial populations. If a conclusive link can be established in healthy horses using faecal material to give an indication of bacterial community structure, then faecal material could potentially become a non-invasive tool to accurately monitor changes in the colonic bacterial populations in response to diet and other environmental factors, and allow for the accurate measurement of potential disease-causing bacteria, such as strains of S. bovis (and other bacteria) in the colon. In human subjects, reports have suggested a potential relationship between increased faecal carrier levels of S. bovis and human gastrointestinal disease(Reference Dubrow, Edberg, Wikfors, Callan, Troncalle, Vender, Brand and Yapp31–Reference Harley33). If a similar trend can be established in the equine, faecal material has the potential to be employed as a model for identifying and monitoring the level of S. bovis in the hindgut, thus detecting rising levels or imbalances at the early stages of disease when treatment can be more effective.
Acknowledgements
The authors are grateful to Cheshire Equine Services for providing hindgut luminal contents. The present study was funded by the Royal (Dick) School of Veterinary Studies, University of Edinburgh. The authors have no conflicts of interest that affect the content or publication of this paper. P. M. H. designed, optimised and validated the real-time PCR assays, K. M. was a University of Edinburgh postgraduate student who carried out aspects of the study, whilst J. M. D. M. secured funding for this research and was responsible for completing some of the real-time PCR assays.






