



Introduction
Papilledema is a common reason for referral to neuro-ophthalmology clinic and typically manifests with bilateral optic disc edema and normal or near-normal afferent visual function. Idiopathic intracranial hypertension (IIH) is the most common cause of papilledema in young women with elevated body mass index.Reference Radhakrishnan, Thacker, Bohlaga, Maloo and Gerryo1 However, even patients fitting this typical demographic should undergo neuroimaging in the form of an MRI brain with venography (MRV). This helps to confirm the diagnosis of raised intracranial pressure (ICP) through visualization of indirect neuroimaging signs of increased ICP. The most sensitive of these is stenosis of the dural venous sinuses, present in nearly all patients.Reference Bidot, Saindane, Peragallo, Bruce, Newman and Biousse2 Venography also allows identification of critical nonidiopathic causes of elevated ICP such as dural venous sinus thrombosis (DVST).
DVST is an important cause of increased ICP and was identified in 11% of patients with the presumed initial diagnosis of IIH in one large series.Reference Agarwal, Kumar and Arora3 In addition to papilledema, DVST can present with a wide range of signs and symptoms, including headache, altered sensorium, and seizures.Reference Ferro, Correia, Pontes, Baptista and Pita4 It can be associated with high morbidity and even mortality due to associated cerebral edema and venous infarction thus all patients with DVST require a thorough work-up in order to identify risk factors predisposing them to thrombotic events.Reference Bousser and Ferro5,Reference Stam6
We reviewed all cases of patients with papilledema seen over 3 years who were found to have DVST and identified four patients amongst this group who were found to have Janus Kinase 2 (JAK2) V617F mutation on subsequent genetic testing. This mutation is well known as a cause of myeloproliferative neoplasms (MPN)Reference James, Ugo and le Couédic7 and in two out of four patients described here it was the presenting event leading to diagnosis of polycythemia vera (PV). These cases illustrate the importance of including JAK2 mutation in the work-up for hereditary causes of hypercoagulability as it has implications for treatment, future thrombotic risk, and long-term prognosis.
Methods
A retrospective chart review was performed of all patients seen in a single tertiary neuro-ophthalmology practice between July 2017 and July 2021 with papilledema who were found to have DVST on MRI of the brain with venography. Clinical charts, results of neuroimaging and results of hypercoagulability work-up were reviewed, including genetic testing for JAK2 mutation. The study protocol was reviewed and approved by the Health Sciences Research Ethics Board at the University of Toronto.
Results
Fifteen patients with papilledema secondary to DVST were included in the study. Of these, 5 (33%) were male. The age of included patients ranged from 19 to 72. All underwent investigations for underlying prothrombotic factors and findings are presented in Table 1. One patient was positive for Factor V Leiden and 3 had systemic conditions that are linked to a hypercoagulable state: pregnancy, Crohn’s disease and Behcet’s disease. In 1 case, a meningioma resulting in severe compression of the left transverse sinus was determined to be the cause of DVST. Four patients (27%) had positive JAK2 V617F mutation on genetic testing. Only 2 out of 15 patients were deemed to have idiopathic DVST after extensive investigations.
Table 1: Cases of dural venous sinus thrombosis seen in neuro-ophthalmology clinic
ND = not done.
Case Descriptions of Patients with DVST Secondary to JAK2 V617F
Case 1
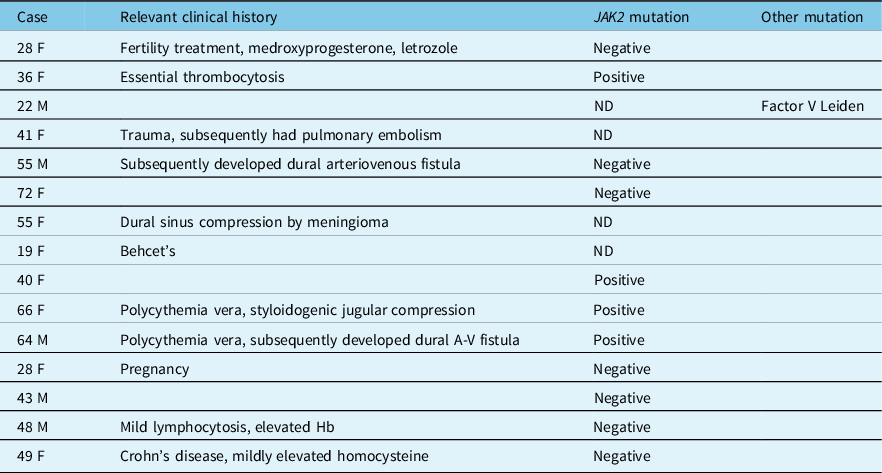
A 40-year-old woman with a history of IIH diagnosed elsewhere 4 years ago presented with a 3-week history of binocular horizontal diplopia, intense headaches, pulsatile tinnitus, nausea, and vomiting. Visual acuity was 20/20 in each eye with no relative afferent pupillary defect (RAPD). She had normal ocular motility with a small esophoria at distance. Dilated fundus exam (DFE) revealed severe bilateral optic nerve edema (Figure 1A), which was corroborated on optical coherence tomography (OCT) of the peripapillary retinal nerve fiber layer (RNFL). 24–2 Humphrey visual field testing was normal in both eyes. MRI and MRV of the brain revealed a thin, nonocclusive linear filling defect in the superior sagittal sinus (Figure 1C, D). Her previous imaging from 4 years earlier was reviewed and no thrombosis was present at this time. A standardized hypercoagulability work-up was undertaken which included peripheral blood assays for antithrombin III, prothrombin mutation, Protein C and S, Factor V Leiden, homocysteine, antiphospholipid, and anticardiolipin antibodies, JAK2, and CALR (calreticulin) mutation. She was found to be positive for the JAK2 V617F mutation. There was no evidence of PV and while she had a history of previously treated anemia, her hemoglobin was within normal limits at 133 g/L. She was started on preventative anticoagulation therapy with warfarin and remained well with no evidence of MPN.
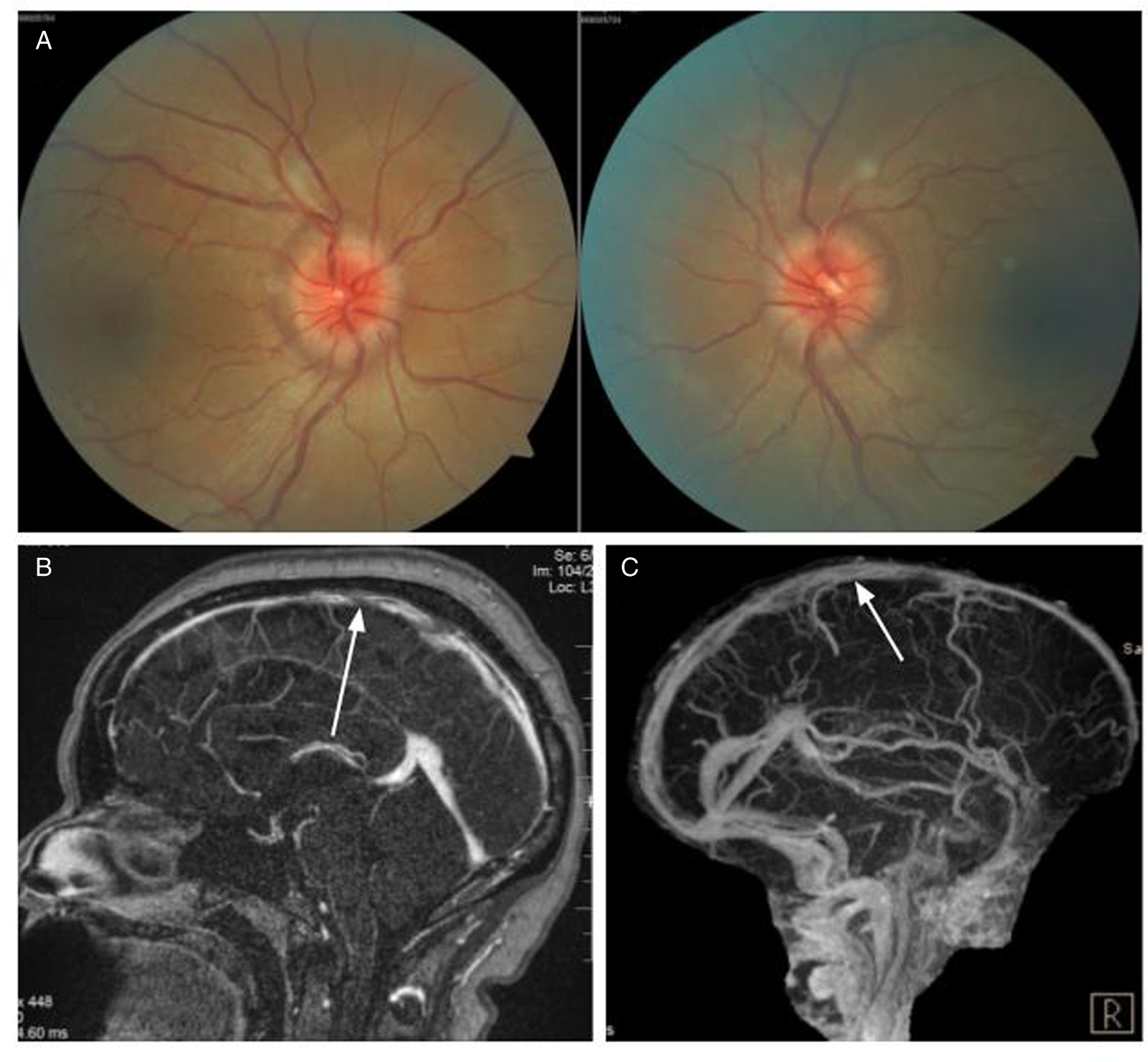
Figure 1: Case 1. (A) Papilledema on fundus examination. (B) MRI and MR venogram (C) showing a nonocclusive thin, linear filling defect in the superior sagittal sinus (white arrows).
Case 2
A 64-year-old man was seen for incidentally discovered bilateral optic disc edema which was not present on his annual optometry screening examination a year ago. A noncontrast computed tomography (CT) head in the emergency room followed by an MRI of the brain were interpreted as unremarkable. He did not have headaches, diplopia, pulsatile tinnitus, or transient visual obstructions. His vision was 20/20 in each eye without an RAPD. DFE revealed severely edematous optic nerves bilaterally (Figure 2A). 24–2 Humphrey visual fields were normal and OCT of the peripapillary RNFL confirmed severe elevation of the optic nerve head in each eye.
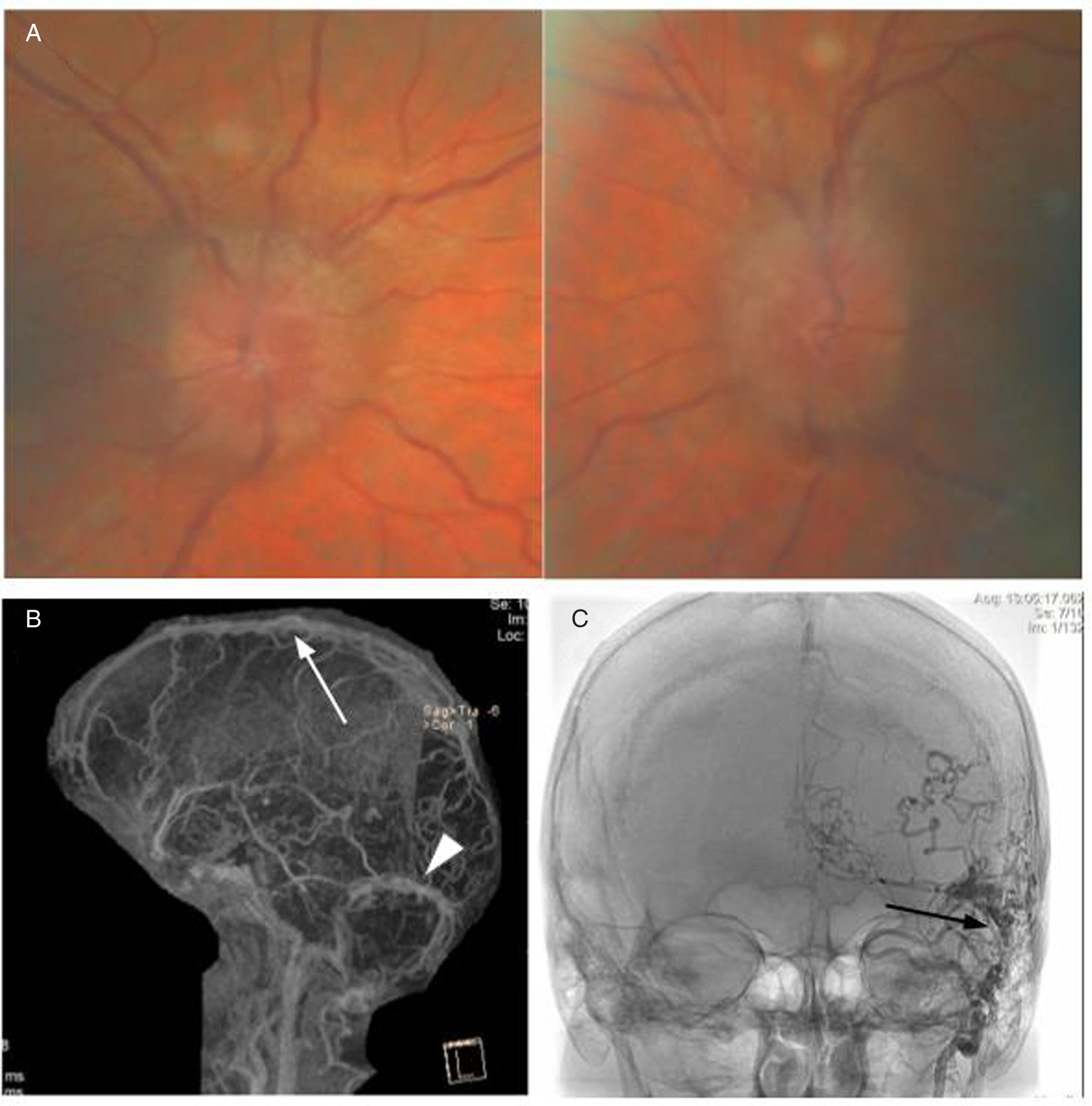
Figure 2: Case 2. (A) Papilledema on fundus examination. (B) MR venogram showing large thrombotic segments of the posterior superior sagittal sinus (white arrow) and left transverse dural venous sinus (white arrowhead). (C) Digital subtraction angiography revealed a left occipital transverse sinus dural arteriovenous fistula (black arrow).
An urgent MRV of the brain revealed extensive thrombosis of the posterior superior sagittal sinus and left transverse sinus (Figure 2B). Numerous serpiginous flow voids along the left transverse sinus and a prominent occipital vessel were suspicious for dural arteriovenous fistula (dAVF). Left occipital-transverse sinus dAVF was confirmed on MR angiography and subsequent digital subtraction angiography (Figure 2C). His hypercoagulability work-up revealed a borderline high hemoglobin (170 g/L) with elevated hematocrit (0.51) and positive JAK2 V617F mutation. Bone marrow biopsy was performed and demonstrated cellular marrow with trilineage hematopoiesis, consistent with the diagnosis of PV. His dAVF was successfully embolized, and he was started on anticoagulation therapy with rivaroxaban. Papilledema improved and he was maintained on cytoreductive treatment with hydroxyurea and repeat phlebotomies.
Case 3
A 66-year-old woman noticed blurry vision in each eye, headache, and pulsatile tinnitus. After multiple visits to an emergency department where she was repeatedly diagnosed with migranous headache, she was referred to an ophthalmologist who noticed bilateral optic disc edema (Figure 3A). CT of the head with CT venography was performed and revealed right transverse and sigmoid sinus thrombosis extending to the internal jugular vein as well as short segment nonocclusive thrombosis of the left distal sigmoid sinus. When assessed in our clinic, vision was 20/25 in each eye with no RAPD. DFE demonstrated bilateral severe optic disc edema with peripapillary choroidal folds. 24–2 Humphrey visual fields demonstrated enlarged blind spot in the right eye and nonspecific changes in the left eye. MRV revealed complete opacification of the right transverse and sigmoid sinus extending through the jugular foramen just below the skull base within the internal jugular vein (Figure 3B, C), with venous stenosis confirmed on angiography (Figure 3D, E). A hypercoagulability work-up was positive for the JAK2 V617F mutation. She also had elevated hematocrit (0.52) and hemoglobin of 168 g/L. Bone marrow biopsy showed atypical megakaryocytes and loose clusters, hypercellularity, and panmyelosis with minimal fibrosis interpreted as consistent with the diagnosis of PV. She was treated with acetazolamide and started on anticoagulation with apixaban and repeat phlebotomies but declined treatment with hydroxyurea. Clinically, she remained stable with no further thrombotic events.
Figure 3: Case 3. (A) Papilledema on fundus examination. (B, C) MRI with venography revealed complete lack of opacification of the right transverse and sigmoid sinus (B, white arrow) extending through the jugular foramen with multiple visible thrombi including within the left jugular vein (C, black arrow). A-P (D) and lateral (E) digital subtraction angiography images showing stenosis of the right transverse and sigmoid sinuses (D, white arrowhead) and the left jugular bulb (D, E, black arrowhead).
Case 4
A 36-year-old woman presented with a 1-year history of increasing headaches and recent onset of blurry vision in each eye. She had been diagnosed with essential thrombocytosis 11 years earlier after incidentally noted elevated platelet counts. Therapy with 81 mg of daily aspirin was recommended by a hematologist; however, she did not adhere to treatment. Her optometrist noted bilateral optic disc edema and referred her to an ophthalmologist, who ordered a CT of the brain with contrast. While awaiting this appointment, she developed nausea and vomiting and went to an emergency department where a noncontrast CT head was normal. Her vision was 20/20 in each eye without an RAPD. DFE revealed severe optic nerve head edema. 24–2 Humphrey visual fields were normal in each eye. Urgent MRI of the brain with venography demonstrated extensive right-sided DVST (Figure 4). Genetic testing was positive for JAK2 V617F mutation. Treatment with warfarin resulted in complete resolution of optic nerve head edema after 2 months.

Figure 4: Case 4. (A) MRI brain with time-of-flight venography showed signal loss in the right sigmoid sinus extending into the jugular vein (white arrowheads). CT venogram coronal (B) and axial (C) confirmed extensive filling defects within transverse and sigmoid sinuses, extending to the jugular vein (white arrows).
Discussion
DVST produces papilledema in the majority of symptomatic cases. In addition to symptoms of increased ICP, patients may present with focal neurologic signs, seizures, or altered level of consciousness. DVST should be suspected in all patients with papilledema as it can present in a very similar manner to IIH as was seen in Case 1.Reference Agarwal, Kumar and Arora3,Reference Eliseeva, Serova, Yakovlev, Mikeladze, Arkhangelskaya and Gasparyan8 All cases of DVST should be investigated for an underlying hypercoagulable state, which is likely to be present in most patients. We found that only 2 out of 15 patients in our series (13%) had no identifiable prothrombotic factors. Increased risk for DVST can be seen with any systemic cause of hypercoagulable state such as pregnancy, hyperhomocysteinemia, antiphospholipid antibodies, myeloproliferative disorders, or other malignancy.Reference Green, Styles and Russell9 There were two patients with DVST in our study who had an underlying systemic inflammatory condition: 1 had a history of Behcet’s disease and 1 had known inflammatory bowel disease, with DVST having been previously reported as a rare complication of both.Reference Uluduz, Midi and Duman10,Reference Cognat, Crassard, Denier, Vahedi and Bousser11 The mechanism of DVST in Behcet’s disease is thought to relate to vasculitis involving dural veins causing phlebitis, while in Crohn’s disease indirect effects on platelets and fibrinolysis are hypothesized to be the causative prothrombotic factors.Reference Danese, Papa, Saibeni, Repici, Malesci and Vecchi12
In the absence of an identifiable prothrombotic cause, genetic predisposition to thrombotic events should be explored. Hereditary causes have been reported in up to 41% of patients with DVST and include Protein C and S deficiency, antithrombin III deficiency, prothrombin mutation, and Factor V Leiden.Reference Silvis, Middeldorp, Zuurbier, Cannegieter and Coutinho13,Reference Komro and Findakly14 The importance of JAK2 mutation as a cause of DVST has only been recognized recently thus we wanted to investigate its prevalence in our cohort of patients with papilledema who were discovered to have DVST and found that it accounted for a full quarter of all DVST cases. While one of the patients with JAK2 V617F mutation had a known diagnosis of MPN, in two patients detection of this mutation led to the diagnosis of MPN.
The JAK2 substitution mutation of phenylalanine (F) for valine (V) at the 617 position was first described in 2005 as a driver mutation in MPN.Reference James, Ugo and le Couédic7 JAK2 V617F mutations and PV are important contributors to venous thrombosis throughout the body, including portal vein thrombosis, deep vein thrombosis, and DVST.Reference de Stefano, Fiorini and Rossi15,Reference Shetty, Kulkarni, Pai, Mukundan, Kasatkar and Ghosh16 In reported series of patients with DSVT, this mutation is found in 6.6%–11% of cases and 60% of these had a myeloproliferative disorder.Reference de Stefano, Fiorini and Rossi15,Reference Passamonti, Biguzzi and Cazzola17 When only patients without myeloproliferative disorder were studied, JAK2 V617F was present in around 3%–6% of DVST cases; these patients tended to be older and have higher, though still normal, hemoglobin and platelet counts.Reference Shetty, Kulkarni, Pai, Mukundan, Kasatkar and Ghosh16,Reference Mahe, Pedersen and Çolak18,Reference Lamy, Palazzo and Agius19 Importantly, a higher risk of recurrent thrombosis has been identified in patients with JAK2 V617F mutation, regardless of the presence of overt myeloproliferative disorder.Reference Lamy, Palazzo and Agius19,Reference Martinelli, de Stefano and Carobbio20
In our series, the prevalence of JAK2 V617F was 26%, which is higher than previously reported, however our sample size is quite small. Our study also included only a subset of all cases of DVST, specifically ambulatory patients with papilledema and though it is unclear why this mutation could be more common in these cases, we speculate that thrombosis in these patients could be more chronic or low grade. Papilledema takes time to develop, and onset is often observed well after the diagnosis of DVST.Reference Liu, Bhatti, John and Chen21 Patients with acute, severe thrombosis would be more likely to present with seizure, altered level of consciousness, and other neurologic symptoms and thus would not be included in our cohort.
DVST is treated with anticoagulation, which may be in the form of heparin, vitamin K antagonists, or novel oral anticoagulants. If there is incomplete recanalization of the thrombus, fibrinolytic agents, mechanical thrombolysis, or thrombectomy may be required.Reference Bose, Graveline, Yogendrakumar, Fergusson and Dowlatshahi22,Reference Saposnik, Barinagarrementeria and Brown23 There is no clear consensus as to the optimal duration of anticoagulation in DVST, with or without JAK2 V617F.Reference Ferro, Bousser and Canhão24
In DVST with JAK2 V617F mutation and no identified MPN, CBC must be monitored on a regular basis. Patients with PV and venous thrombotic events require anticoagulation in addition to specific therapy for their myeloproliferative disease, the mainstay of which consists of cytoreductive therapy with phlebotomy and antineoplastic agents such as hydroxyurea.Reference de Stefano, Za and Rossi25,Reference Marchioli, Finazzi and Specchia26 Timely administration of hydroxyurea in patients with PV can reduce the number of thrombotic events and greatly increases median survival time in symptomatic disease.Reference Tefferi, Guglielmelli and Larson27,Reference Chievitz and Thiede28
Conclusion
DVST is an important secondary cause of papilledema seen in the ophthalmology clinic. We propose that genetic testing including JAK2 mutation analysis should be performed in all patients with DVST with an abnormal CBC, absence of risk factors for thrombosis or when hypercoagulable work-up is unrevealing. Testing for this allele is readily available in tertiary centers as it is widely performed in the evaluation of myeloproliferative disorders. Ophthalmologists should be aware of the JAK2 V617F mutation due to this increased risk of myeloproliferative disease and risk of recurrent thrombosis and these patients should be comanaged with hematologists. Early diagnosis and treatment in these patients can dramatically improve outcomes.
Conflicts of Interest
The authors have no conflict of interest to report.
Statement of Authorship
LD and EM designed the study, LD and AD collected data and wrote the manuscript, EM revised the manuscript for intellectual content, all authors approved the final draft.





