Infantile-onset multisystem neurologic, endocrine, and pancreatic disease (IMNEPD) has recently been described.Reference Hu, Matter and Issa-Jahns1, Reference Picker-Minh, Mignot and Doummar2 The index case involved two patients from a consanguineous family of Yazidian–Turkish descent with a homozygous nonsense mutation in PTRH2. The core clinical features include postnatal microcephaly, delayed speech and motor development with regression, intellectual disability, sensorineural hearing loss, progressive ataxia, cerebellar atrophy, peripheral sensorimotor neuropathy, and dysfunction of the pancreas, thyroid, and liver of varying severity.Reference Hu, Matter and Issa-Jahns1, Reference Picker-Minh, Mignot and Doummar2, Reference Sharkia, Shaley and Zalan3
Herein, we report three IMNEPD patients from a consanguineous family of Syrian descent. The three affected children (patient III-6, patient III-3, patient III-2; Figure 1) are from healthy parents who are first cousins. There are three unaffected children in the family who are 18 months, 23 years, and 26 years old (Figure 1).
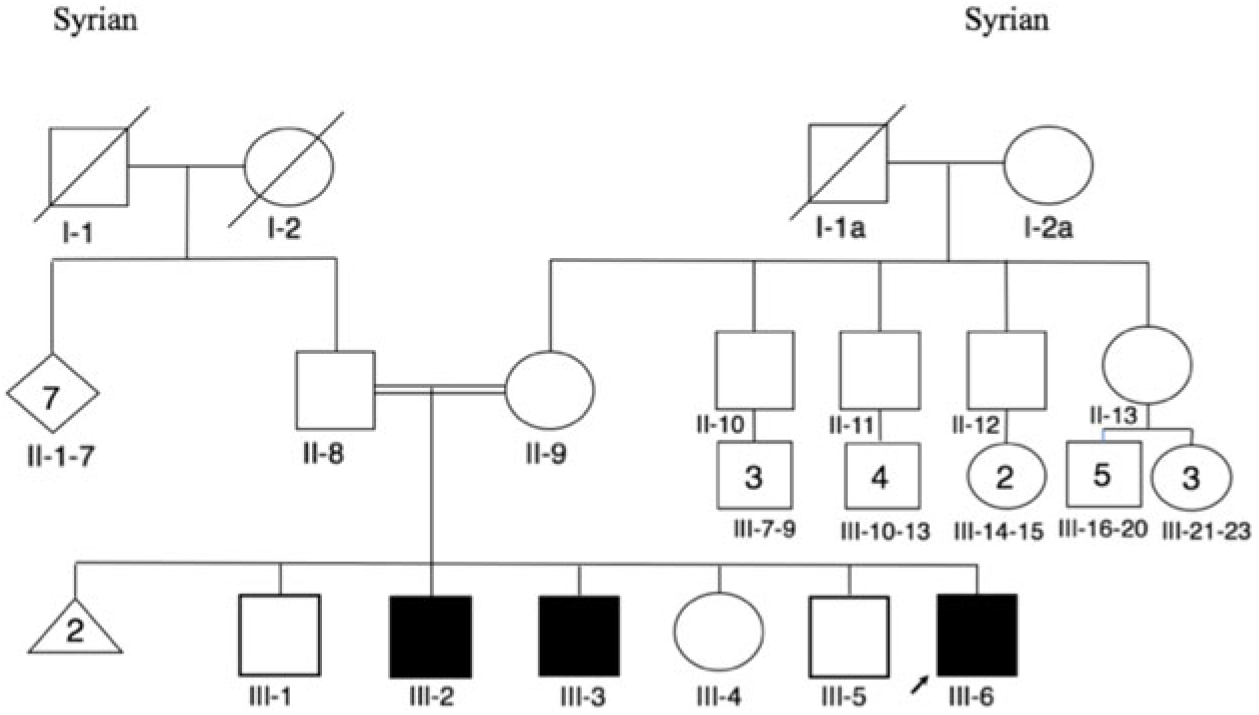
Figure 1: Pedigree.
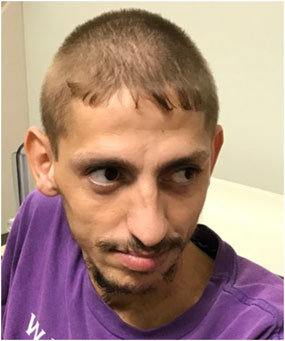
Patient III-6 is 27 years old and is most severely affected. He had a hypoxic birth event. He first spoke at the age of 4, and regressed within 1 year to nonspecific vocalizations. He lost independent ambulation at 14 months as he developed progressive truncal and limb ataxia. He has distal, more than proximal, weakness and is wheelchair-dependent. He has flexion contractures of his hands and feet, and high foot arches. At 7 years, he developed generalized tonic clonic and myoclonic seizures. He has postnatal microcephaly, mild bilateral ptosis, exotropia, mild facial dysmorphisms with hypertelorism, and a thin upper lip vermillion (Figure 2). He was diagnosed with insulin-dependent diabetes mellitus (IDDM) and microcytic anemia due to iron deficiency at age 18 and seronegative spondyloarthropathy at age 26.
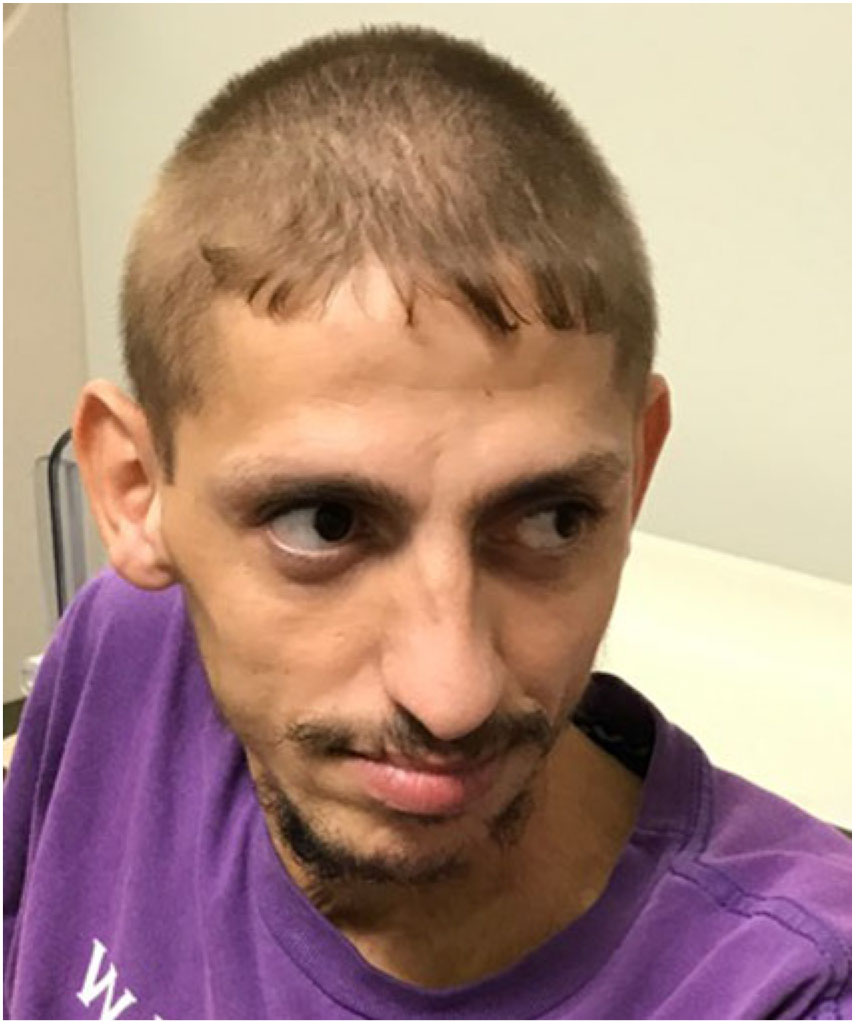
Figure 2: Patient III-6 with microcephaly, mild bilateral ptosis, exotropia, hypertelorism, and thin upper lip vermillion.
Patient III-3 is 17 years old. He had an uncomplicated birth, but displayed postnatal microcephaly, motor and language regression at 1 year. He has progressive truncal and limb ataxia and a broad-based, high-steppage gait and requires a walker. He has a limited vocabulary of less than five words. He has mild bilateral ptosis, global symmetric hypotonia, hyporeflexia with distal muscle wasting that is more significant in the upper than lower extremities. He was diagnosed with IDDM at age 16 and has suspected myoclonic seizures.
Patient III-2 is 7 years old and has postnatal microcephaly with developmental regression. He acquired a limited vocabulary at the age of 2. He is treated with clonidine for hyperactivity. He could walk independently by the age of 3 and has a broad-based gait with mild truncal ataxia and hip girdle weakness with no muscle wasting. He is currently being investigated for nocturnal frontal lobe seizures and myoclonus. He has an undescended right testis. He does not have pancreatic dysfunction, thyroid, or liver disease.
Initial investigations for patient III-6 revealed an elevated random glucose 12 mmol/L (normal 3.4–11) but normal glycated hemoglobin, liver enzymes, and homocysteine levels. Serum biotinidase assay, urine organic acids, plasma and urine amino acids, and serum cholesterol were all within normal limits, while X-linked adrenoleukodystrophy testing was excluded as the plasma very-long-chain fatty acids were normal. Hepatitis B and C serology was negative. A nerve conduction study was performed and revealed a sensorimotor axonal and demyelinating polyneuropathy. Electromyography was not consistent with a primary myopathy.
Cranial magnetic resonance imaging (MRI) revealed symmetrical, bilateral cerebellar atrophy in the two eldest affected patients (Figure 3). The brainstem, corpus callosum, and cerebral cortex were within normal limits. Cranial MRI in the youngest affected sibling was normal. Electroencephalograms (EEG) for patients III-2 and III-3 demonstrated background slowing with increased asynchronous delta activity consistent with diffuse encephalopathy without epileptiform activity.
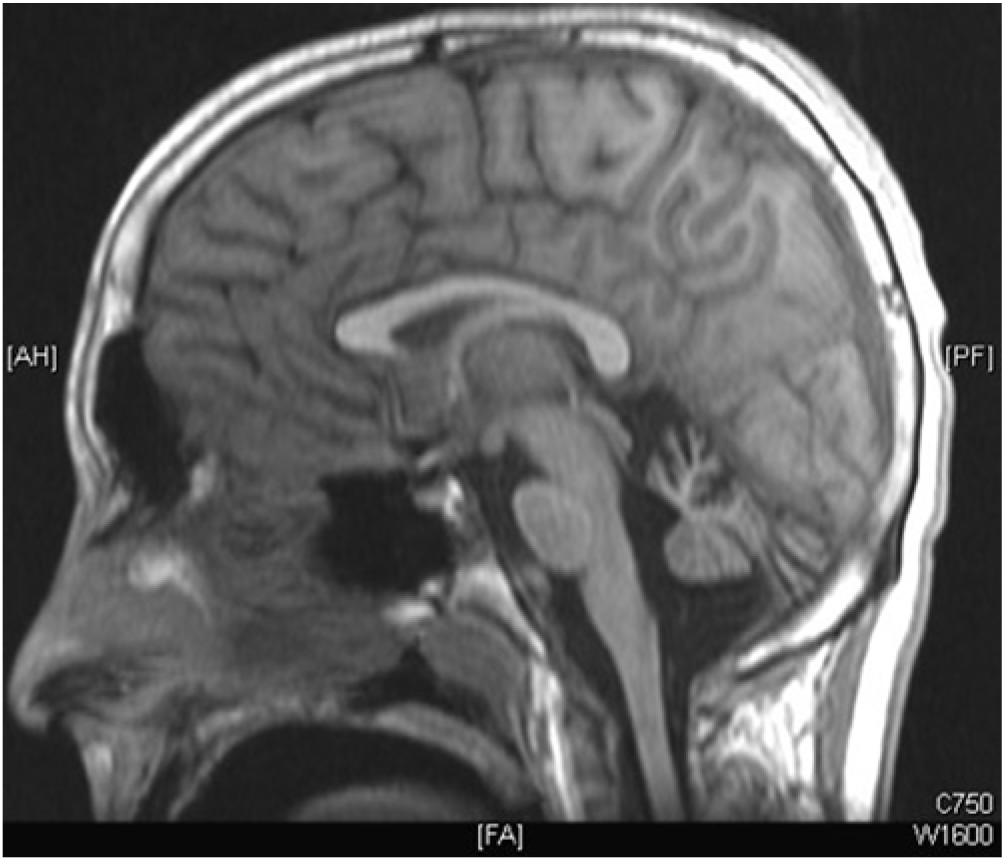
Figure 3: MRI brain T1 sagittal; prominent cerebellar atrophy.
The history of consanguinity suggested an autosomal recessive pattern of inheritance. In the context of middle eastern background, shared clinical features, and multisystemic involvement, a causative mutation in the mitochondrial protein PTRH2 was suspected. All exons of PTRH2 were PCR-amplified and sequenced bidirectionally. All three brothers had a homozygous nonsense mutation in PTRH2, c.324G>A (p.W108*). This G-to-A mutation results in a premature stop at codon p.108, truncating the protein by 71 amino acids and removing most of the amino acids that participate in the enzymatic active site.Reference Jan, Matter and Pai5 The c.324G>A (p.W108*) mutation is not listed in the human Gene Mutation Database Cardiff (NCBI dbSNP, 1000 Genomes, Exome Variant Server or ClinVar). This very rare variant has been identified with an allele frequency of 0.000004063 in the gnomAD database with one heterozygous individual in 246114 alleles (all databases accessed January 22, 2018). Clinical phenotyping was used exclusively to focus the genetic analysis. We had a high index of suspicion for IMNEPD, and we have confirmed the same pathogenic variant in all three siblings and thus have not pursued whole-exome sequencing. To our knowledge, there is no relationship to any other known patients; however, we recognize that given the consanguinity there is a possibility for more than one autosomal recessive condition to affect this family.
PTRH2 (peptidyl-tRNA hydrolase 2) is a highly conserved mitochondrial protein encoded on chromosome 17. The substrates of PTRH2 may be peptidyl-tRNAs that dissociate from ribosomes during incomplete protein synthesis.Reference Hu, Matter and Issa-Jahns1 During protein translation, a significant proportion of the ribosomes that initiate the mRNA readout do not reach the stop codon. Peptidyl-tRNA molecules may dissociate from the mRNA template and terminate the process prematurely.Reference Hu, Matter and Issa-Jahns1, Reference Picker-Minh, Mignot and Doummar2, Reference Sharkia, Shaley and Zalan3, Reference De Pereda, Waas, Jan, Ruoslahti, Schimmel and Pascual4 In addition, PTRH2 also acts as a phosphoprotein that regulates NFkB and ERK signaling and participates in integrin-mediated apoptotic signaling and survival. When a cell attaches to the extracellular matrix, PTRH2 interacts with focal adhesion kinase and activates the PI3K–AKT–NFkB pathway to enhance attachment.Reference Hu, Matter and Issa-Jahns1, Reference Jan, Matter and Pai5 Upon loss of cell attachment, PTRH2 is phosphorylated, released into the cytosol, and combines with transcriptional regulator amino-terminal enhancer of split, which triggers apoptosis or anoikis.Reference Hu, Matter and Issa-Jahns1, Reference Jan, Matter and Pai5 PTRH2 is expressed in the neurons of the neocortex, basal ganglia, hippocampus and cerebellum, testis, prostate, skeletal muscle, liver, and, to a lesser extent, in the heart, spleen, placenta, and colon.Reference Hu, Matter and Issa-Jahns1
Murine models of homozygous Ptrh2-mutant mice developed postnatal dystrophy and died within 2 weeks of life.Reference Hu, Matter and Issa-Jahns1 Ptrh2-knockout mice were normal at birth, but demonstrated progressive decrease in length and weight, muscle wasting, ataxia, and reduced brain weight with cerebellar atrophy.Reference Hu, Matter and Issa-Jahns1 These mice also demonstrated exocrine pancreatic insufficiency.Reference Hu, Matter and Issa-Jahns1
A review of the literature was conducted using search terms of IMNEPD in OMIM and PubMed. Consistent with previous descriptions, our three patients displayed the core features of IMNEPD, including postnatal microcephaly, motor and speech delay, intellectual disability, ataxia, peripheral neuropathy, and seizures with varying severity (Table 1). The two older brothers also have pancreatic insufficiency. None of our patients have evidence of thyroid or liver dysfunction (Table 1). To date, patients with IMNEPD have been described with variable dysmorphisms, including midface hypoplasia, hypertelorism, thin upper lip vermilion, proximally placed thumbs, and deformities of the fingers and toes.Reference Hu, Matter and Issa-Jahns1, Reference Alazami, Patel and Monies6 Recently, a milder phenotypic variant characterized by mild microcephaly, peripheral sensorimotor neuropathy, sensorineural hearing loss, and delayed puberty was described in three sisters who shared a homozygous PTRH2 missense mutation c.254A>C (p.Q85P).Reference Sharkia, Shaley and Zalan3
Table 1: Clinical characteristics of patients with IMNEPD
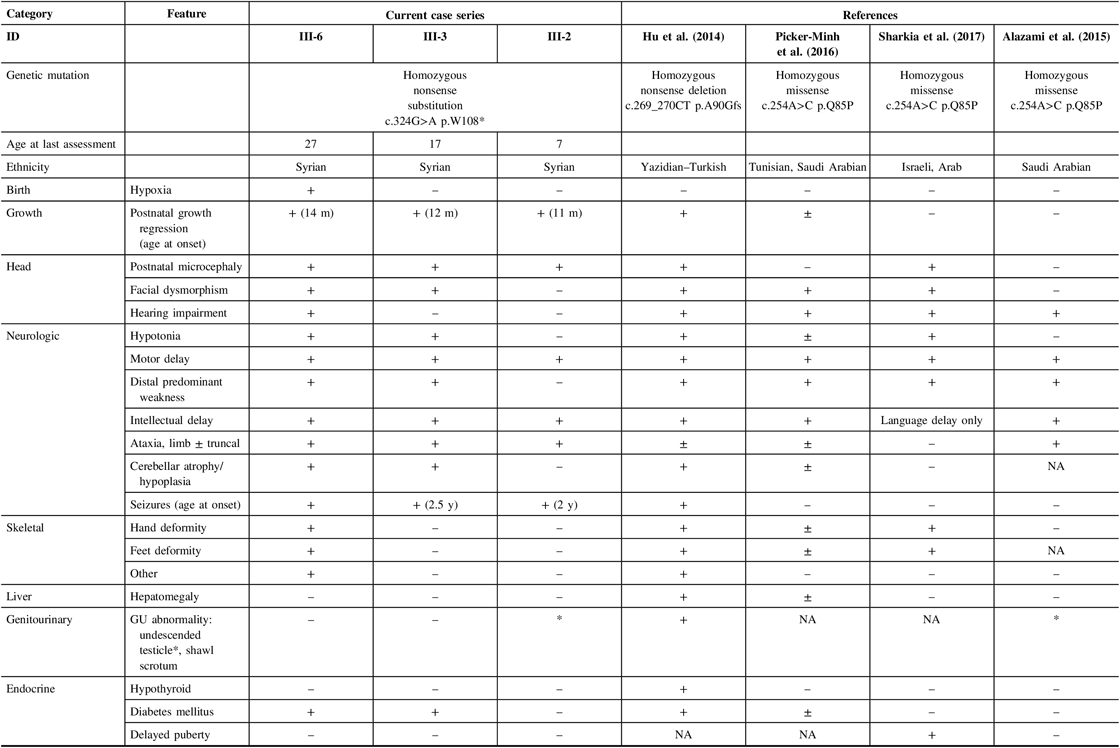
In our patients, we identified a novel, causal homozygous nonsense G-to-A mutation in PTRH2 at nucleotide c.324. In review of the previously described mutations, it appears that nonsense mutations of PTRH2, which produce a truncated protein, may result in a more severe phenotype characterized by prominent microcephaly, motor and speech delay, intellectual disability, cerebellar atrophy, and pancreatic dysfunction. With missense mutations, there is likely more preserved function in the altered PTRH2 leading to a milder phenotype. In the present family, however, we observed phenotypic variability between siblings with the same mutation suggesting variable expressivity for PTRH2 mutations. Future studies will help to determine the degree to which puberty is affected in this disorder and help to validate the observed phenotypic variants of PTRH2 mutations.
In patients with suspected IMNEPD, the multisystemic features should yield a wide differential diagnosis, including mitochondrial disorders. Patients may have overlapping features with syndrome of short stature, microcephaly, and endocrine dysfunction; Johanson–Blizzard syndrome; progressive myoclonic epilepsies; spinocerebellar ataxias; or specific metabolic disorders.Reference Hudson and Chinnery7, Reference Cohen, Chinnery, Copeland, Adam, Ardinger and Pagon8, Reference de Bruin, Mericq and Andrew9
Initial investigations for patients with suspected IMNEPD should include complete blood count, biochemical assays for electrolytes, creatinine, urea, creatine kinase, thyroid-stimulating hormone, B12, glucose, glycated hemoglobin, lactate, liver function enzymes, ammonia, plasma amino acids, and urine organic acids. Additional testing may include nerve conduction study, electromyography, cranial MRI, and EEG. Genetic testing of the parents and siblings is important to determine the risk of recurrence and carrier status, and age-appropriate counselling would be necessary for all affected patients and siblings.
At present, there is no specific treatment for this condition; however, the related complications of this disorder can be addressed. In the context of seizures, valproic acid toxicity may occur given that IMNEPD is a mitochondrial cytopathy, and therefore, other antiepileptic medications should be used. We believe that patients would benefit from a collaborative care plan with multidisciplinary physicians and allied health practitioners.
Acknowledgements
Written informed consent was obtained for the genetic analysis, publication of data, and imaging. We are grateful to Wendy McCaul, BSc, for performing mutation analysis, and to Melanie Napier (genetic counsellor) for assistance with genetic counselling.
Abbreviations
FAK, focal adhesion kinase; Pi3K, phosphatidylinositol 3-kinase protein kinase B; AKT, protein kinase B; NFkB, nuclear factor kappa light chain enhancer of activated B cells; ERK, extracellular signal-related kinase; mTOR, mammalian target of rapamycin; pS6, phosphorylated protein S6; ESR, erythrocyte sedimentation rate; CRP, C-reactive protein, POLG, DNA polymerase subunit gamma.
Electronic Database Information
URLs used in the preparation of this article are Locus Link, National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), 1999. https://www.ncbi.nlm.nih.gov/pubmed
Online Mendelian Inheritance in Man, OMIM™, Center for Medical Genetics, Johns Hopkins University (Baltimore, MD) and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), 1999. http://www.ncbi.nlm.nih.gov/omim/
Disclosures
The authors have no conflicts of interest to declare.
Statement of Authorship
All author contributed equally to the manuscript.