



Introduction
The recommendations below provide an overview of best practices as of 2024 in the diagnosis of select tumours of the central nervous system including diffuse and circumscribed gliomas, glioneuronal tumours, ependymomas and medulloblastomas. Reference Louis, Perry and Wesseling1–Reference Farrell, Shi, Bodman and Olson8 These recommendations have been reviewed and approved at a national level by the Canadian Association of Neuropathologists (CANP). This document is expected to evolve over time in response to continued scientific study and clinical experience.
The document also serves to describe the minimum expectations for Canadian diagnostic laboratories to support the diagnosis of brain tumours to the current standards of the WHO, including the immunohistochemical, molecular and cytogenetic tests detailed below. For optimal workflow and timely patient care, it is recommended that decentralised testing be maximised where possible, although methylation profiling may require a more centralised approach.
The document was produced through the process described in the Abstract, combining the search for relevant scientific literature and consensus among the CANP members. The authors have been chosen to represent the diverse inputs, including the Professional Affairs Committee (DGM), the Executive Committee (AG), the president (RH) and secretary/treasurer of the association, a resident (MG) and a recognised leader in the field (CH).
Diffuse Gliomas
Diffuse gliomas are divided into adult-type and paediatric-type tumours. Adult-type diffuse gliomas include (1) isocitrate dehydrogenase (IDH)-mutant astrocytomas and oligodendrogliomas and (2) glioblastoma, IDH-wildtype. Paediatric-type diffuse gliomas are divided into (3) low-grade (characterised mainly by alterations in the genes in the MAPK pathway or MYB/MYBL1 genes) and (4) high-grade (characterised by a diverse array of alterations in histones, mismatch repair, receptor tyrosine kinases and others). Reference Brat, Aldape and Bridge2,Reference Zarnett, Sahgal and Gosio9–Reference Rousseau, Bennett and Lim-Fat12 Histologically, diffuse gliomas do not typically show distinctive features, so careful use of immunohistochemical and molecular testing is required to achieve an integrated diagnosis.
In adult patients, adult-type molecular alterations predominate. IDH-mutant gliomas should be distinguished from those that are IDH-wildtype, which have significantly poorer prognoses. Paediatric-type alterations should be considered in younger adults. Reference Brat, Aldape and Bridge2,Reference Lim-Fat, Macdonald and Lapointe5,Reference Zarnett, Sahgal and Gosio9,Reference Rousseau, Bennett and Lim-Fat12
Please see Figure 1 for an adapted diagnostic flow chart for diffuse gliomas.
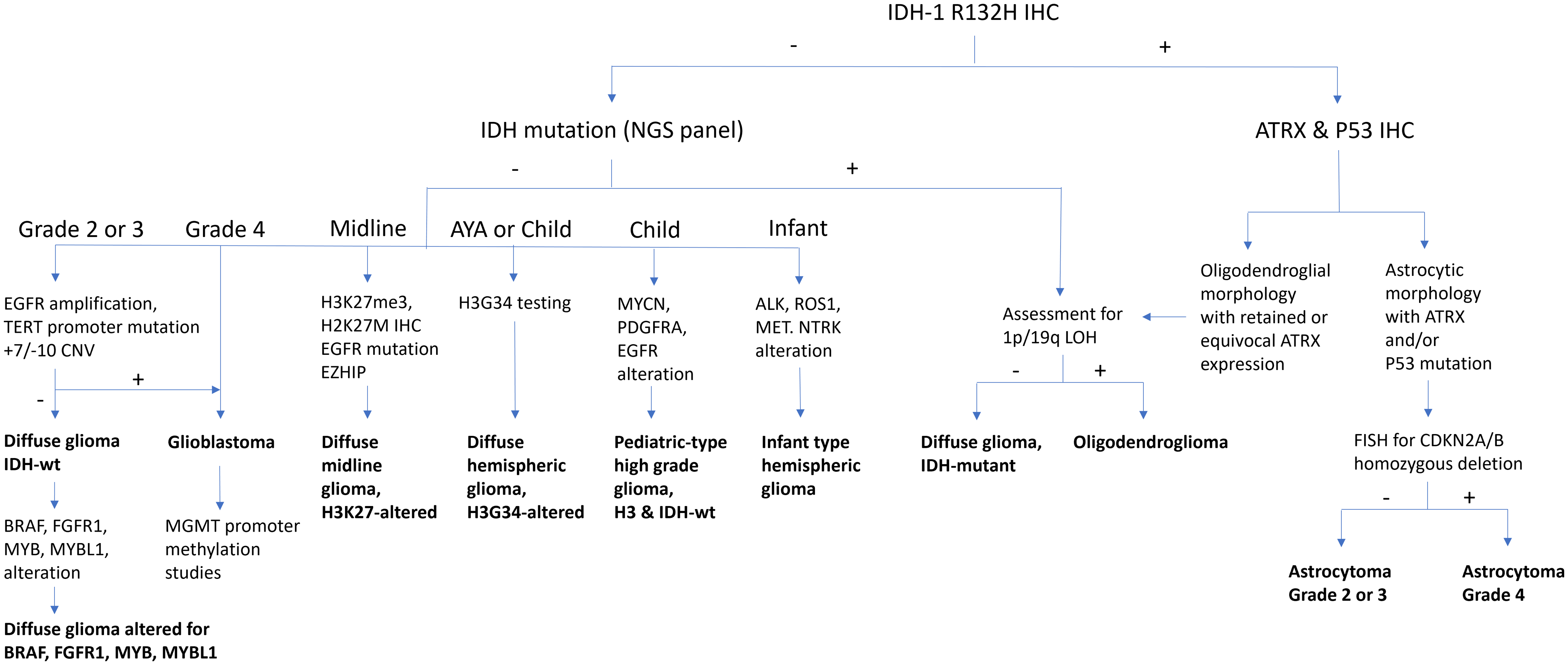
Figure 1. Diffuse glioma diagnostic algorithm. 1p/19q = short arm of chromosome 1 and long arm of chromosome 19; ALK = anaplastic lymphoma kinase; ATRX = alpha-thalassaemia mental retardation X-linked; BRAF = v-raf murine sarcoma viral oncogene homologue B1; CDKN2A/B = cyclin-dependent kinase inhibitor 2A/B; CNV = copy number variants; EGFR = epidermal growth factor receptor; EZHIP = enhancer of zeste inhibitory protein; FGFR1 = fibroblast growth factor receptor 1; FISH = fluorescence in situ hybridisation; H3G34 = histone H3 mutation at codon 34; H3K27M = histone H3 mutation at codon 27; H3K27me3 = histone H3 trimethylation at lysine 27; IDH = isocitrate dehydrogenase; IHC = immunohistochemistry; LOH = loss of heterozygosity; MET = mesenchymal–epithelial transition; MGMT = O (6)-methylguanine-DNA-methyltransferase; MYB = myeloblastosis transcription factor; MYBL1 = MYB proto-oncogene like 1; MYCN = myelocytomatosis viral oncogene neuroblastoma-derived homologue; NGS = next-generation sequencing; NTRK1 = neurotrophic tyrosine kinase receptor; P53 = transformation-related protein 53; PDGFRA = platelet-derived growth factor receptor alpha; R132H = mutation in codon 132 of IDH1; ROS1 = c-ros oncogene 1; TERT = telomerase reverse transcriptase; wt = wildtype.
-
1. The most common mutation in IDH1 (IDH1 p.R132H) may be assessed by immunohistochemistry (IHC). It can be combined with IHC for ATRX ± p53. In the context of an IDH mutation, ATRX loss (usually accompanied by aberrant p53 expression) is diagnostic of astrocytoma. Retained ATRX expression, on the other hand, should prompt testing for 1p/19q-codeletion, which is diagnostic of oligodendroglioma. The copy number status of CDKN2A/B may be assessed for grading of histologically lower-grade IDH-mutant astrocytomas. CDKN2A/B homozygous deletion has been linked to reduced survival and may serve as a molecular marker of grade 3 in oligodendrogliomas as well. Reference Louis, Perry and Wesseling1,Reference Brat, Aldape and Bridge2,Reference Rousseau, Bennett and Lim-Fat12–Reference Galbraith, Garcia and Wei14
-
2. Tumours negative for IDH1 p.R132H may be assessed further depending on the clinical context.
-
i. In patients over the age of 55 years with a tumour showing glioblastoma histology, further diagnostic testing is generally unnecessary (glioblastoma, IDH-wildtype). Reference Zarnett, Sahgal and Gosio9,Reference Yuen, Barbaro and Haggiagi11
-
ii. In patients under the age of 55 years and/or with a tumour showing lower-grade histology, the tumour should be further assessed for “non-canonical” IDH1 and IDH2 mutations, and as appropriate, molecular alterations associated with glioblastoma, IDH-wildtype (see recommendation 3) and/or paediatric-type alterations (see recommendations 5, 6 and 7) should also be considered. Reference Berger, Wen, Lang-Orsini and Chukwueke15,Reference Mohile, Messersmith and Gatson16
-
-
3. Histologically, lower-grade IDH-wildtype diffuse gliomas, especially in those arising in middle-aged or older adults, should be tested for molecular alterations of glioblastoma, IDH-wildtype: combined chromosome Ch7 gain/Ch10 loss, epidermal growth factor receptor (EGFR) amplification and/or telomerase reverse transcriptase (TERT) promoter mutation. Reference Brat, Aldape and Bridge2,Reference Berzero, Di Stefano and Ronchi17–Reference Capper, Reifenberger and French18
-
4. MGMT promoter methylation should be assessed in all glioblastoma, IDH-wildtype. It is at present unclear if IDH-mutant astrocytoma patients would benefit from this test. Reference Lim-Fat, Macdonald and Lapointe5,Reference Farrell, Shi, Bodman and Olson8,Reference Melhem, Detsky, Lim-Fat and Perry19
In children and younger adults, diffuse gliomas should be tested for paediatric-type alterations (see recommendations 6, 7 and 8). In paediatric and young adult patients, paediatric-type alterations are common and amenable to targeted therapy. Adult-type alterations (IDH1, IDH2) should be considered in patients of adolescent age and older. The location of the tumour (hemispheric vs. midline) also plays an important role in guiding testing.
-
5. Hemispheric diffuse low-grade gliomas should be assessed for alterations (SNVs, indels, fusions) in the MAPK pathway (including FGFR1, FGFR2, FGFR3, KRAS, NF1, BRAF), MYB and MYBL1. Less common alterations in NTRK1, NTRK2, NTRK3, MAP2K1 and MET may be included. Adult-type alterations should be considered. BRAF p.V600E can be assessed by IHC. BRAF p.V600E-mutant tumours should be assessed for CDKN2A/B copy number status. CDKN2A/B homozygous deletion and/or TERT and/or ATRX alterations should prompt consideration of an alternative diagnosis (see below: Circumscribed gliomas and glioneuronal tumours, recommendation 2). Reference Lim-Fat, Macdonald and Lapointe5–Reference Stone, Mankad and Tan7,Reference Jonsson, Lin and Young13,Reference Galbraith, Garcia and Wei14
-
6. Hemispheric diffuse high-grade gliomas should be assessed for mutations in H3-3A (diffuse hemispheric glioma, H3G34-mutant), SNVs and amplifications in EGFR and PDGFRA, alterations in genes involved in cancer predisposition syndromes (mismatch and replication repair [MLH1, MSH2, MSH6, PMS2, POLE, POLD1], TP53) and MYCN amplification. Adult-type alterations should be considered. Reference Lim-Fat, Macdonald and Lapointe5,Reference Stone, Mankad and Tan7,Reference Khuong-Quang, Buczkowicz and Rakopoulos20,Reference Schwartzentruber, Korshunov and Liu21 There may be histologic overlap with tumours currently classified as “circumscribed” (see below: Circumscribed gliomas and glioneuronal tumours, recommendation 2). DNA methylation profiling may serve to distinguish between entities with overlapping molecular features. H3 p.G34R and MMR may be assessed by IHC. In infants, fusions involving receptor tyrosine kinases including ALK, ROS1, NTRK1, NTRK2, NTRK3 and MET should be assessed (infant-type hemispheric glioma). Reference Lim-Fat, Macdonald and Lapointe5,Reference Rudà, Capper and Waldman22
-
7. Diffuse gliomas of the midline (thalamus, brainstem, cerebellum, spinal cord) in patients of all ages should be assessed for H3K27 trimethylation (H3K27me3) by IHC and for alterations in H3-3A (or less commonly, H3C2, H3C3 and H3C14). H3 p.K28M (K27M) can be assessed by IHC. Secondary alterations in BRAF or FGFR1 may be included. EGFR amplification and enhancer of zeste inhibitory protein (EZHIP) overexpression (IHC) should be assessed in H3-wildtype cases. Reference Khuong-Quang, Buczkowicz and Rakopoulos20–Reference Rudà, Capper and Waldman22
Circumscribed Gliomas and Glioneuronal Tumours
Most circumscribed gliomas and glioneuronal tumours are characterised by alterations (SNVs, indels, fusions) in the MAPK pathway. Copy number alterations in CDKN2A/B and alterations in genes in telomere maintenance (ATRX, TERT) are important additional alterations in particular tumour types.
Rare tumours in this category harbour specific molecular alterations. Reference Galbraith, Garcia and Wei14,Reference Rudà, Capper and Waldman22,Reference Sahm, Brandner and Bertero23
-
1. Pilocytic astrocytoma should be assessed for alterations in the MAPK pathway, including BRAF (SNVs and fusions), FGFR1 (SNVs, fusions, internal tandem duplication) and NF1. Testing may include less commonly altered genes such as KRAS, PTPN11 and RAF1.
-
2. High-grade astrocytoma with piloid features (HGAP) and pleomorphic xanthoastrocytoma (PXA) should be assessed for alterations in CDKN2A/B (copy number loss) and ATRX and/or TERT, in addition to MAPK pathway alterations (BRAF, FGFR1, NF1). A matching DNA methylation profile is an essential diagnostic criterion for HGAP and desirable for PXA. HGAP can be highly favoured in the setting of RAS/MAPK alteration plus ATRX loss and/or CDKN2A/B homozygous deletion; however, definitive diagnosis currently requires methylation profiling.
-
3. Ganglioglioma should be assessed for alterations in BRAF. BRAF p.V600E may be assessed by IHC. Less commonly, alterations in other MAPK pathway genes including RAF1, KRAS and NF1 may be included. CDKN2A/B homozygous deletion should be absent.
-
4. The diagnosis of a variety of rare circumscribed gliomas and glioneuronal tumours may be confirmed by assessing for particular molecular alterations or by a specific DNA methylation profile. In these cases, next-generation sequencing is preferred over methylation profiling as it may provide the specific target for therapy.
-
• Dysembryoplastic neuroepithelial tumour: FGFR1 SNVs, fusions, ITD
-
• Papillary glioneuronal tumour: PRKCA fusions
-
• Rosette forming glioneuronal tumour: FGFR1, NF1 and/or PIK3CA alterations
-
• Myxoid glioneuronal tumour: PDGFRA SNVs
-
• Diffuse leptomeningeal tumour: BRAF fusions with 1p loss ± 19q loss ± 1q gain
-
• Multinodular vacuolating neuronal tumour: MAP2K1 SNVs
-
• Extraventricular neurocytoma: FGFR1 fusions
-
• Desmoplastic infantile ganglioglioma/astrocytoma: BRAF, RAF1, FGFR1 alterations
-
• Diffuse glioneuronal tumour with oligodendroglioma-like features and nuclear clusters: distinct DNA methylation profile
-
• Astroblastoma: MN1 fusions
-
• Chordoid glioma: PRKCA SNVs
-
Ependymoma
-
1. Posterior fossa ependymomas should be tested for H3K27me3 loss by IHC, along with EZHIP expression by IHC if possible, to distinguish posterior fossa group A (PFA) from posterior fossa group B (PFB) ependymoma. DNA methylation profiling is an alternative method that may be helpful in adult cases where PFB versus methylation class subependymoma is a more likely differential. PFA ependymoma should be assessed for copy number changes in 1q and 6q. Methylation class subependymoma should be assessed for TERT promoter mutation and chromosome 6 loss. Reference Gillen, Riemondy and Amani24,Reference Northcott, Buchhalter and Morrissy25
-
2. Supratentorial ependymomas should be tested for ZFTA and YAP1 fusions. In those with ZFTA fusion, CDKN2A/B copy number status should be assessed.
-
3. Spinal cord ependymomas with high-grade histology should be assessed for MYCN amplification.
Medulloblastoma
-
1. All medulloblastomas should undergo molecular testing to determine the molecular subgroup. DNA methylation profiling and/or NanoString analysis are suitable techniques. Assessment of copy number alterations may also be indicated depending on subgroup (e.g. MYC amplification in group 3).Reference Northcott, Buchhalter and Morrissy 25
-
2. SHH-activated medulloblastoma in the paediatric age group should be assessed for TP53 alterations.
The tables summarise the molecular alterations (Table 1), crucial immunohistochemical stains (Table 2) and tumour types for which the DNA methylation profile is considered an “essential” diagnostic criterion by WHO (Table 3). In Table 3, we highlight with an asterisk those tumour types for which methylation profiling is the only method to reach a diagnosis. For all other tumour types in this table, next-generation sequencing represents an alternative approach. In addition, any unresolved cases could benefit from methylation analysis. Table 4 summarises the defining molecular alterations in gliomas and glioneuronal tumours.
Table 1. Summary of genes and molecular alterations in central nervous system neoplasms
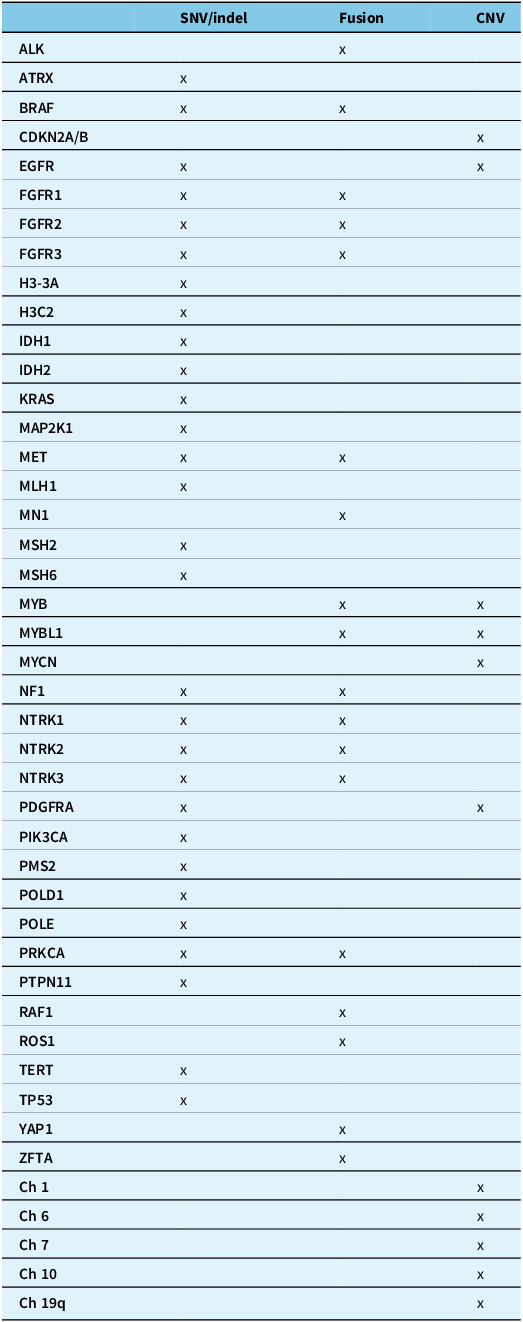
ALK = anaplastic lymphoma kinase; ATRX = alpha-thalassaemia mental retardation X-linked; BRAF = v-raf murine sarcoma viral oncogene homologue B1; CDKN2A/B = cyclin-dependent kinase inhibitor 2A/B; Ch = chromosome; CNV = copy number variants; EGFR = epidermal growth factor receptor; FGFR1/2/3 = fibroblast growth factor receptor 1/2/3; H3-3A = histone H3 variant H3.3; H3C2 = H3 clustered histone 2; IDH1/2 = isocitrate dehydrogenase 1/2; KRAS = Kirsten rat sarcoma viral oncogene homologue; MAPK21 = mitogen-activated protein kinase kinase 1; MET = mesenchymal–epithelial transition; MLH1 = human mutL homologue 1; MN1 = meningioma 1; MSH2/6 = MutS homologue 2/6; MYB = myeloblastosis transcription factor; MYBL1 = MYB proto-oncogene like 1; MYCN = myelocytomatosis viral oncogene neuroblastoma-derived homologue; NF1 = neurofibromatosis type 1; NTRK1/2/3 = neurotrophic tyrosine kinase receptor 1/2/3; PDGFRA = platelet-derived growth factor receptor alpha; PIK3CA = phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; PMS2 = post-meiotic segregation increased 2; POLD1 = DNA polymerase delta 1; POLE = DNA polymerase epsilon; PRKCA = protein kinase C alpha; PTPN11 = tyrosine-protein phosphatase non-receptor type 11; q:long arm of chromosome; RAF1 = rapidly accelerated fibrosarcoma; ROS1 = c-ros oncogene 1; SNV/indel = single nucleotide variant/insertions and deletions; TERT = telomerase reverse transcriptase; TP53 = tumour protein p53; YAP1 = yes-associated protein 1; ZFTA = zinc finger translocation associated.
Table 2. Essential immunohistochemical stains
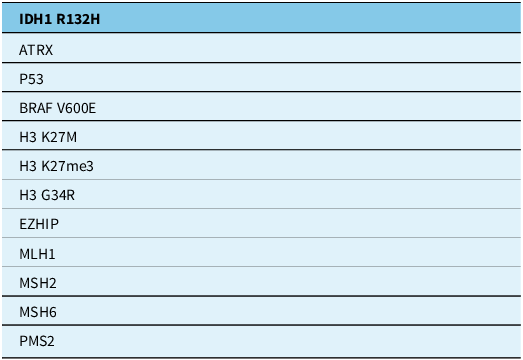
ATRX = alpha-thalassaemia mental retardation X-linked; BRAF V600E = v-raf murine sarcoma viral oncogene homologue B1 mutation in codon 600; H3G34 = histone H3 mutation at codon 34; H3 K27M = histone H3 mutation in codon 27; H3K27me3 = histone H3 trimethylation at lysine 27; IDH1 R132H = isocitrate dehydrogenase 1 mutation at codon 132; MLH1 = human mutL homologue 1; MSH2/6 = MutS homologue 2/6; PMS2 = post-meiotic segregation increased 2; P53 = tumour protein p53.
Table 3. Tumour types in which the DNA methylation profile is included in WHO CNS 5e as an “essential” diagnostic criterion
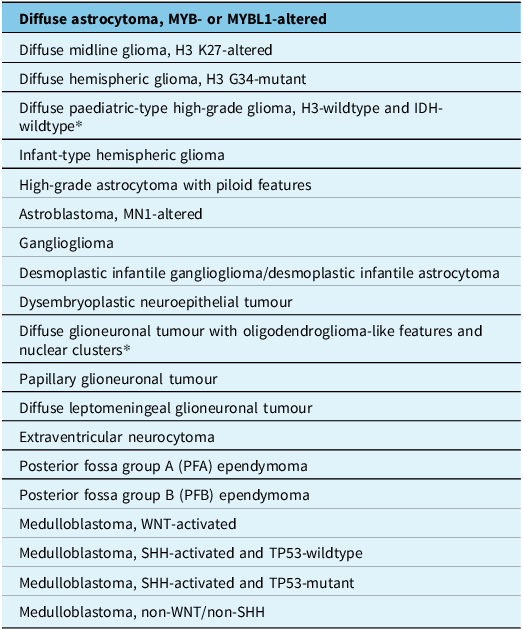
*Asterisks identifies tumor types in which methylation profiling is the only method to reach a diagnosis.
Table 4. Summary of gliomas and glioneuronal tumours
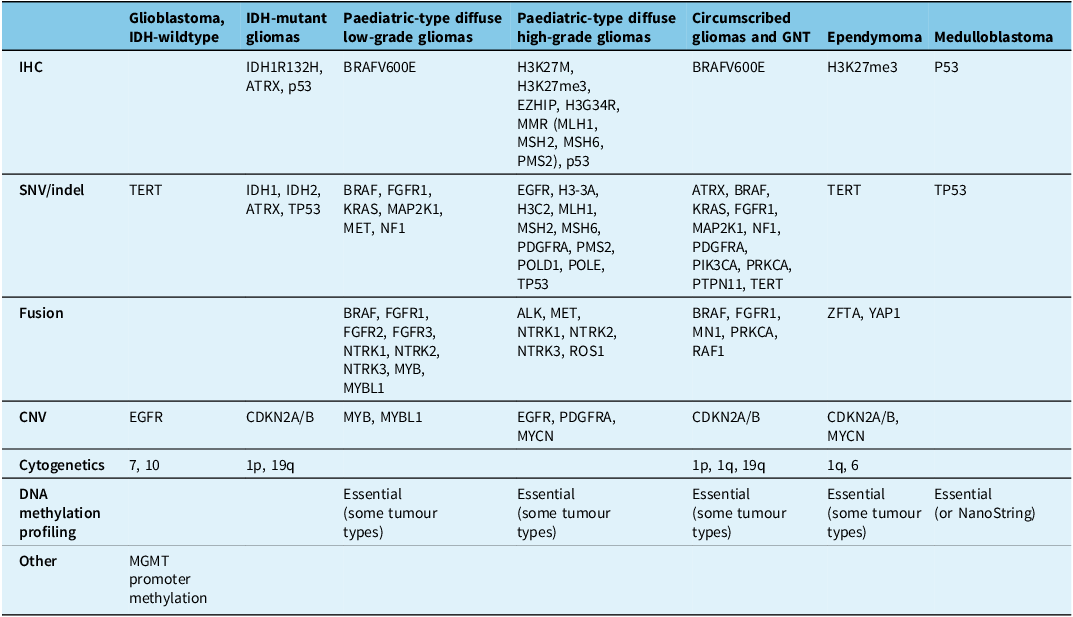
ALK = anaplastic lymphoma kinase; ATRX = alpha-thalassaemia mental retardation X-linked; BRAF = v-raf murine sarcoma viral oncogene homologue B1; CDKN2A/B = cyclin-dependent kinase inhibitor 2A/B; CNV = copy number variants; EGFR = epidermal growth factor receptor; EZHIP = enhancer of zeste inhibitory protein; FGFR1/2/3 = fibroblast growth factor receptor 1/2/3; H3-3A = histone H3 variant H3.3; H3C2 = H3 clustered histone 2; H3G34 = histone H3 mutation at codon 34; H3K27M = histone H3 mutation at codon 27; H3K27me3 = histone H3 trimethylation at lysine 27; IDH 1/2 = isocitrate dehydrogenase 1/2; IHC = immunohistochemistry; MAP2K1 = mitogen-activated protein kinase kinase 1; MET = mesenchymal–epithelial transition; MLH1 = human mutL homologue 1; MN1 = meningioma 1; MSH2/6 = MutS homologue 2/6; MGMT = O (6)-methylguanine-DNA-methyltransferase; MMR = mismatch repair MYB = myeloblastosis transcription factor; MYBL1 = MYB proto-oncogene like 1; MYCN = myelocytomatosis viral oncogene neuroblastoma-derived homologue; NF1 = neurofibromatosis type 1; NTRK1/2/3 = neurotrophic tyrosine kinase receptor; P53 = transformation-related protein 53; p = short arm of a chromosome; q = long arm of a chromosome, PDGFRA = platelet-derived growth factor receptor alpha; PIK3CA = phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; PMS2 = post-meiotic segregation increased 2; POLD1 = DNA polymerase delta 1; POLE = DNA polymerase epsilon; PRKCA = protein kinase C α; PTPN11 = tyrosine-protein phosphatase non-receptor type 11; RAF1 = rapidly accelerated fibrosarcoma; ROS1 = c-ros oncogene 1; TERT = telomerase reverse transcriptase; TP53 = tumour protein p53; YAP1 = yes-associated protein-1; ZFTA = zinc finger translocation associated.
Acknowledgements
The CANP is indebted to other members of the Professional Affairs Committee (Dr Sidney Croul, Dr Marie-Christine Guiot, Dr Jason Karamchandani, Dr Ian Mackenzie, Dr Frank van Landeghem) for leading the development of these recommendations. We also recognise many other members who participated in the discussions, as well as our many international colleagues who have contributed to this evolving body of knowledge for the good of our patients.
Author contributions
All authors have participated in discussions regarding the content and in the preparation of the manuscript. In addition, DGM chaired the Professional Affairs Committee and wrote the first draft, AG extensively edited the manuscript and wrote the algorithm and tables, RH originated the idea of preparing the recommendations and presided over the process, PWS organised the meeting where the recommendations were discussed, MG contributed to the discussion and editing of the manuscript and CH provided general guidance and specific editing.
Funding statement
None.
Competing interests
None.






 Open access
Open access