



Introduction
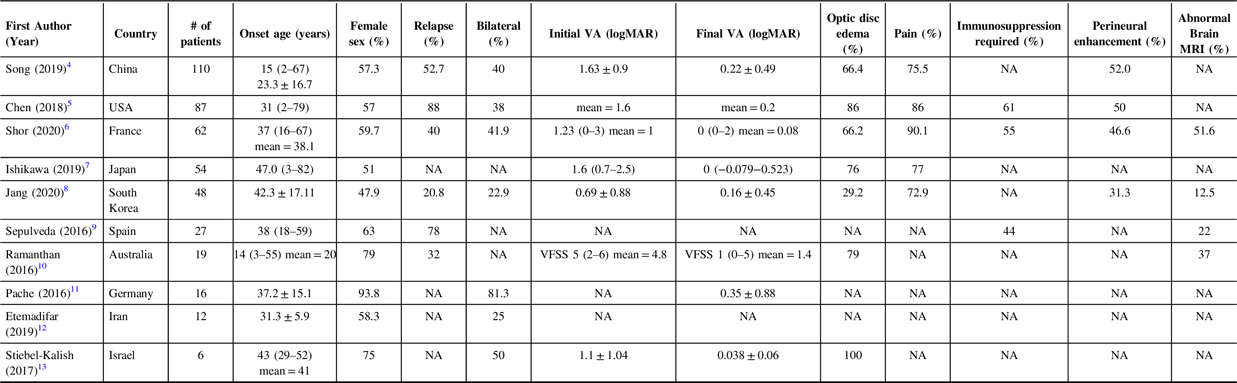
Optic neuritis (ON) is an inflammatory optic neuropathy that typically presents with vision loss and pain with eye movements. Immunoglobulin-G antibodies against myelin oligodendrocyte glycoprotein (MOG-IgG) have been identified in patients with ON, leading to MOG-IgG-associated ON (MOG-IgG ON) being established as a distinct disease. Reference Chun and Cestari1–Reference Cobo-Calvo, Vukusic and Marignier3 Since then, many studies have been conducted to characterize MOG-IgG ON regarding its clinical phenotype, radiological features, treatments, and outcomes. Studies of cohorts of MOG-IgG ON patients have been reported from the United States (US), Iran, Japan, South Korea, Germany, Australia, Spain, France, China, and Israel (Table 1). Reference Song, Zhou, Yang, Xu, Sun and Wei4–Reference Stiebel-Kalish, Lotan and Brody13 However, there is a lack of such research in Canadian patients. To our knowledge, the largest report of Canadian adults with MOG-IgG ON in the literature to date only discussed three patients and did not include optical coherence tomography (OCT), formal visual field data, or comprehensive ophthalmic exam data. Reference Alshamrani, Alnajashi, Shosha, Casserly and Morrow14 An abstract presented at a Canadian conference described 30 patients with MOG-IgG disease but it did not include visual field, OCT, or vision data. Reference Cross, Ackermans and Mattar15 The goal of this study is to contribute to characterization of MOG-IgG ON in a novel Canadian cohort with respect to clinical presentation, radiologic findings, treatment, and outcomes.
Table 1: Summary of Largest Cohort of MOG-IgG ON Patients from Other Countries (data reported as median (range) or mean ± SD; NA = not available in the data reported within the study)
Methods
This was a retrospective review of consecutive patients presenting to tertiary neuro-ophthalmology practices at the University of Toronto with a diagnosis of MOG-IgG ON from July 2017 to June 2020. University of Toronto Research Ethics Board approval was obtained. Patients were included if they had at least one episode of vision loss attributable to MOG-IgG ON. A diagnosis of ON was defined an as inflammatory optic neuropathy meaning that vision loss was attributable to optic nerve dysfunction [reduced visual acuity, visual field defect, reduced color vision, and changes in the optic nerve appearance with support from magnetic resonance imaging (MRI) showing enhancement of the optic nerve if performed within the appropriate time course]. The presence of MOG-IgG was assessed in the serum via a cell-based assay (Mitogen Advanced Diagnostic Lab, Calgary) and the titers were classified as low, medium, or high according to laboratory standards. All patients were evaluated in a standardized fashion by the same two neuro-ophthalmologists (EM, JM) over the study period. The neuro-ophthalmic evaluation included Snellen visual acuity, pupillary examination, formal visual field testing (Humphrey 24-2 SITA-Fast), and OCT (Cirrus HD-OCT 5000, Carl Zeiss Meditec, Dublin, CA) of the retinal nerve fiber layer (RNFL) and ganglion cell-inner plexiform layer (GCIPL). Snellen visual acuity was converted to logMAR visual acuity for analysis. Formal Humphrey visual fields were reviewed for all patients and the mean deviation was recorded. The average OCT RNFL and GCIPL thickness was also recorded for each patient. An ON attack was considered acute if the vision loss occurred within 1 month, subacute if it occurred between 1 and 3 months, and chronic if it occurred beyond 3 months from the initial neuro-ophthalmology visit.
All data was exported into Microsoft Excel and data analysis was performed with the same program. Results were reported as the mean (± standard deviation) or median (range).
Results
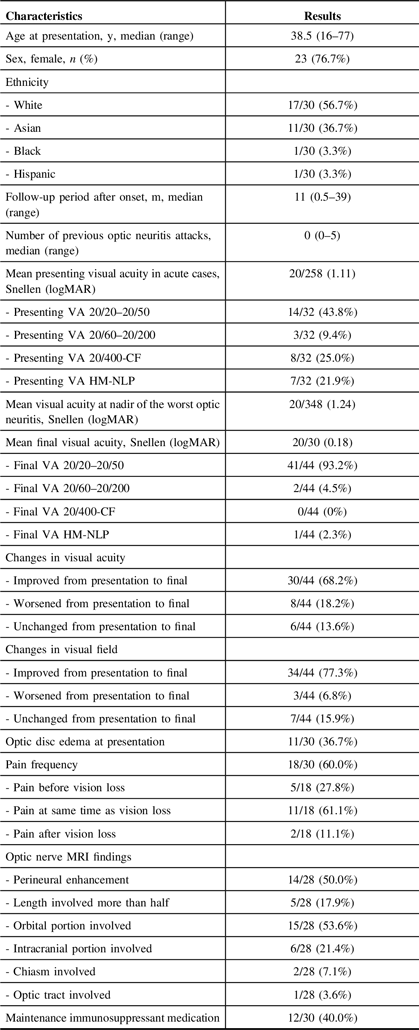
A total of 46 eyes of 30 patients were included in the study (Table 2). Two patients had a history of cancer (ovarian and breast) in remission and one patient had a history of autoimmune disease (Sjogren’s syndrome). The mean age at presentation was 40.7 ± 14.4 years. Most patients had bilateral involvement (16/30; 53.3%) with the right eye only involved in 7 patients and the left eye in 7 patients.
Table 2: Demographic and Clinical Features in MOG Antibody-Positive Patients (n = 30) and Eyes (n = 46) with Optic Neuritis
All patients had MOG-IgG detectable in the serum and the titer was classified as low in 47% of cases, medium in 21% of cases, and high in 32% of cases. All patients were negative for aquaporin-4 IgG and five patients had the presence of MOG-IgG tested at another laboratory (St. Joseph’s Hospital, Toronto, Ontario) with consistent results.
Seventeen (56.7%) patients presented with their first episode of ON rather than a recurrent attack. The mean number of previous ON attacks prior to presentation was 0.90 ± 1.35. Four patients had recurrence after the index visit for this study, none of whom had further attacks once immunosuppression was started.
Nine patients had associated headache. Isolated ON was present in 27 patients and ON was associated with other neurological symptoms in 3 patients. These neurological symptoms included homonymous hemianopia and bilateral lower extremity weakness due to associated transverse myelitis.
Twenty-two patients were seen during an acute episode of ON, three during a subacute phase, and five were seen more than 3 months after an episode of vision loss. For those presenting with acute symptoms, mean duration of vision loss was 13.0 ± 8.1 days and mean duration of pain was 12.1 ± 6.7 days. At acute presentation, mean visual acuity was 20/258 (logMAR 1.11) and mean Humphrey MD was −16.90 ± 10.83 dB. Ten patients seen in the acute period had optic disc edema whereas 2 had a normal appearing optic nerve and 10 had at least some visible pallor of the involved optic nerve. Seven of those patients with pallor had previous episodes of ON. For those with optic disc edema, the average OCT RNFL thickness was 164.23 ± 46.53 um.
Patients with acute vision loss were treated with high-dose corticosteroids in 20 cases. This took the form of intravenous methylprednisolone 1 g daily administered over 5 days in 17 cases and oral prednisone at a dose of 1250 mg for 5 days in 3 cases. Fourteen patients were given a tapering dose of oral prednisone after initial treatment with high dose prednisone and five were treated with plasma exchange (PLEX) due to poor response to high-dose corticosteroids. Nine patients with acute vision loss were started on long-term immunosuppression with mycophenolate mofetil (n = 3), azathioprine (n = 3), cyclophosphamide (n = 1), or rituximab (n = 2). The reason for immunosuppression was severe attack with incomplete recovery (n = 3) and recurrent attacks (n = 6).
Patients seen during the subacute or chronic period had an average visual acuity of 20/49 (logMAR 0.39) at presentation (range 20/20 to NLP). For patients with previous history of ON not seen during an acute period, the most recent episode occurred 8.28 ± 11.82 months (with a range from 5 weeks to 3 years) before the date of the initial neuro-ophthalmology visit.
An MRI of the brain and orbits were performed in all patients. Nineteen patients had an MRI of the brain and orbits within 1 month of vision loss. Orbital MRI in these patients showed perineural enhancement in 11 patients with more than half the length of the optic nerve involved in 4. The orbital portion was involved in 11, intracranial portion in 5, optic chiasm in 2, and optic tract in 1. A spine MRI was performed in 15 patients and was normal in 11 and showed abnormalities in 4. Representative examples with the optic nerve appearance are shown in Figure 1. The abnormalities were described as follows: demyelinating lesions in the upper thoracic cord; confluent cord signal and swelling of the thoracic cord; longitudinally extensive cord lesions in the cervical spine; and thin, elongated T2-hyperintensity in the cervicomedullary junction along with elongated, expansile intramedullary T2-hyperintensity from T1 to T3. An MRI of the brain was normal in almost all patients (28/30) with two patients having additional intracranial lesions. These included T2/FLAIR hyperintensities in the left midbrain, pons, medulla, frontal, and parietal lobes.
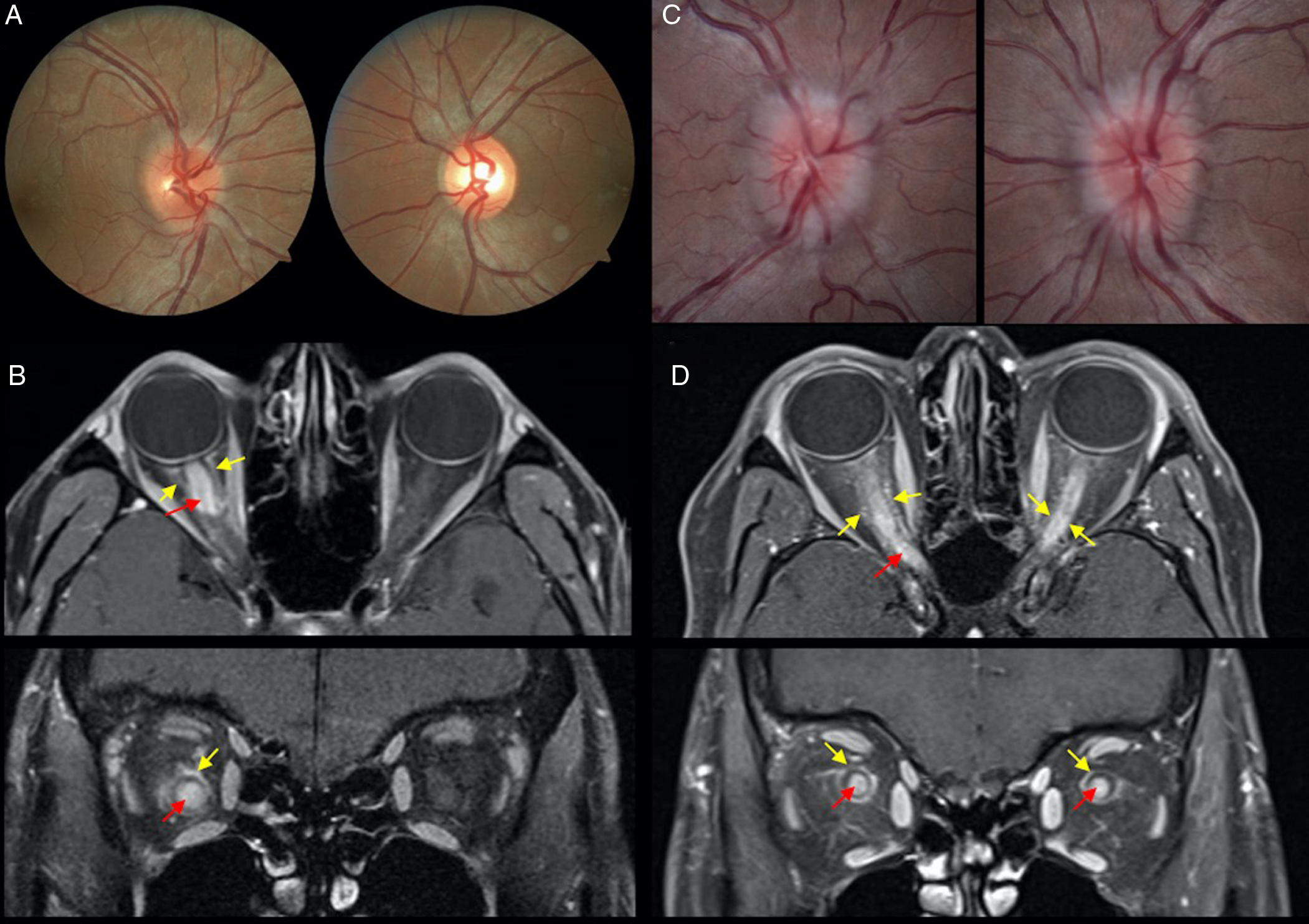
Figure 1: A case of right optic neuritis in a 29-year-old Asian man with counting fingers visual acuity at presentation and mild right optic disc edema (A). Magnetic resonance imaging (MRI) orbits with fat suppression post-contrast shows optic nerve (red arrow) and optic nerve sheath enhancement (yellow arrow; B). A case of bilateral optic neuritis in a 34-year-old woman with visual acuity of counting fingers (right eye) and hand motions (left eye) with bilateral optic disc edema (C). There is optic nerve (red arrow) and optic nerve sheath (yellow arrow) enhancement on MRI orbits with fat suppression post-contrast (D).
Mean duration of follow-up for all 30 patients was 14.45 ± 11.59 months. Final visual acuity was 20/30 (logMAR 0.18), Humphrey mean deviation was −7.17 ± 8.85 dB, OCT RNFL thickness was 72.15 ± 20.16 um, and OCT GCIPL was 62.60 ± 11.58 um.
Discussion
Our cohort of 30 patients with MOG-IgG ON demonstrated a female predominance and older age at presentation compared to typical ON. Reference Beck16 Most patients were Caucasian. Just over half of patients reported eye pain or pain with eye movements and most cases were isolated without other neurological symptoms. Most patients had a normal MRI of the brain, consistent with international guidelines that suggest testing for MOG-IgG in patients with a normal brain MRI. Reference Jarius, Paul and Aktas17 In patients with acute presentation, visual acuity was often worse than 20/200 with over one-fifth of these patients demonstrating incomplete recovery after treatment with high-dose corticosteroids and thus additionally treated with PLEX. Long-term immunosuppression was eventually initiated in 40% of patients and most demonstrated good final visual outcome with 20/30 average acuity at last follow-up.
The demographics of patients with MOG-IgG ON remain a topic of debate as previous studies included different patient populations and different geographic locations. Most patients in our cohort were female. Many previous studies have found a somewhat equal sex distribution, reporting a range between 47.9% and 63% females in their cohorts, with a few showing significant female predominance (Table 1). Reference Song, Zhou, Yang, Xu, Sun and Wei4–Reference Stiebel-Kalish, Lotan and Brody13 The highest reported percentages of women were 93.8% in a German study of 16 patients and 79% from in an Australian study of 19 patients. Reference Ramanathan, Prelog and Barnes10,Reference Pache, Zimmermann and Mikolajczak11 No studies have shown a predominantly male cohort, with the highest reported percentage of males being 52.1% in a Korean cohort. Reference Jang, Kim and Yun8 The age of onset of MOG-IgG ON can be variable as some studies have reported average onset in the second, Reference Song, Zhou, Yang, Xu, Sun and Wei4,Reference Ramanathan, Prelog and Barnes10 fourth, Reference Chen, Flanagan and Jitprapaikulsan5,Reference Shor, Aboab and Maillart6,Reference Sepulveda, Armangue and Martinez-Hernandez9,Reference Pache, Zimmermann and Mikolajczak11,Reference Etemadifar, Abbasi, Salari, Etemadifar and Tavakoli12 or fifth decade of life. Reference Ishikawa, Kezuka and Shikishima7,Reference Jang, Kim and Yun8,Reference Stiebel-Kalish, Lotan and Brody13 Our patients ranged in age from 16 to 77 with a mean age of 40.7 years, which is consistent with the previously reported notion that MOG-IgG ON can affect people across a wide range of ages. Reference Chen and Tariq Bhatti2 This information is important as MOG-IgG ON should be kept in the differential diagnosis for all patients presenting with ON and age is not helpful in ruling out this condition.
Many studies reported bilateral eye involvement in around half of their cases of MOG-IgG ON, which is consistent with our cohort. Reference Song, Zhou, Yang, Xu, Sun and Wei4–Reference Shor, Aboab and Maillart6,Reference Stiebel-Kalish, Lotan and Brody13 The visual acuity of patients with acute presentations was significantly reduced, consistent with other studies reporting severe impairment of visual acuity at onset and nadir. Reference Song, Zhou, Yang, Xu, Sun and Wei4–Reference Ishikawa, Kezuka and Shikishima7,Reference Stiebel-Kalish, Lotan and Brody13 The MOG-IgG titer – whether low, medium, or high – did not seem to influence initial visual acuity. This supports the idea that the antibody titer does not influence the clinical phenotype. Reference Chen and Tariq Bhatti2 Regarding optic disc appearance, our patients were less likely to have edema than patients in most other cohorts, which reported rates of edema as high as 86% to 100%. Reference Chen, Flanagan and Jitprapaikulsan5,Reference Stiebel-Kalish, Lotan and Brody13 The higher proportion of optic disc pallor in our acute cases could largely be accounted for by the previous episodes of ON in these cases. Despite the poor vision at presentation, most patients had a good visual outcome and less than 5% of patients had a final visual acuity of 20/200 or worse, which has been reported to range from 6% to 14% of patients. Reference Chen and Tariq Bhatti2,Reference Song, Zhou, Yang, Xu, Sun and Wei4,Reference Chen, Flanagan and Jitprapaikulsan5 The good final visual acuity was consistent with the preserved peripapillary OCT RNFL thickness in our group (72.15 um), which was better compared to previous reports from Germany (59 um) and China (68.7 um). Reference Song, Zhou, Yang, Xu, Sun and Wei4,Reference Pache, Zimmermann and Mikolajczak11
Pain or pain with eye movements was common, headache was seen in a minority of individuals and neurological issues were infrequent in our patients. Pain is a frequent symptom reported in previous cohorts and can range from 75% to 86% of patients. Reference Song, Zhou, Yang, Xu, Sun and Wei4–Reference Ishikawa, Kezuka and Shikishima7 Headache occurrence has been reported to be as high as 50%. Reference Asseyer, Hamblin and Messina18 Associated neurological symptoms were rare in our patients, but this may simply be a reflection of our patients being derived from neuro-ophthalmology practices. Transverse myelitis has been previously reported to be as high as 36% to 50% in previous cohorts of ON patients. Reference Chen, Flanagan and Jitprapaikulsan5,Reference Pache, Zimmermann and Mikolajczak11 Perineural enhancement on MRI was common in our group of patients, consistent with previous literature that has reported this finding in 31.3%–52.0% of patients. Reference Song, Zhou, Yang, Xu, Sun and Wei4–Reference Shor, Aboab and Maillart6,Reference Jang, Kim and Yun8,Reference Etemadifar, Abbasi, Salari, Etemadifar and Tavakoli12 The rare optic chiasm (7.1%) and tract (3.6%) involvement are congruent with previous reports. Reference Song, Zhou, Yang, Xu, Sun and Wei4–Reference Shor, Aboab and Maillart6 The large majority of patients with MRI of the spine were normal and it was rare for patients to have abnormal spinal imaging when the brain was normal. Over 90% of our patients had a normal MRI of the brain, alerting the clinicians that normal brain MRI in patients presenting with ON should increase the suspicion for MOG-IgG associated disease.
There is a lack of consensus regarding optimal acute treatment for MOG-IgG ON attacks and long-term management to prevent additional attacks. Reference Chen and Tariq Bhatti2 Intravenous methylprednisolone was the most used first-line agent for acute vision loss in our cohort, but prednisone 1250 mg was also used in a few cases. All but one patient had at least some improvement with high-dose corticosteroids, supporting this as a reasonable first-line treatment for acute MOG-IgG attacks. As demonstrated in our cohort, PLEX can be considered in patients who do not have significant improvement with high-dose corticosteroids, as this is an antibody-mediated disease. Reference Chen and Tariq Bhatti2 No patients were given IVIG in our cohort, due to difficulty in accessing this agent in Ontario. Long-term immunosuppression was used in 40.0% of patients in our series. None of these patients had a relapse after being started on immunosuppressive therapy, supporting rituximab, mycophenolate mofetil, and azathioprine as reasonable chronic immunosuppressive agents in MOG-IgG ON. Reference Chen and Tariq Bhatti2 Only 13.3% (4/30) of patients in our cohort had relapses after their initial visit, with one patient having more than one relapse. Other studies with established follow-up periods have reported relapses in at least 32% and up to 88% of their patients, making our relapse rate much lower. However, our follow-up time of 14.45 months was shorter than those studies, so it is possible that more patients in our cohort will have relapses in the future. Reference Song, Zhou, Yang, Xu, Sun and Wei4–Reference Shor, Aboab and Maillart6,Reference Ramanathan, Prelog and Barnes10 A recent multicenter retrospective study found that the lowest annualized relapse rate was associated with maintenance IVIG therapy, but larger studies are needed to determine the comparative effectiveness of each. Reference Chen, Flanagan and Bhatti19
The strengths of our study include a relatively large cohort of MOG-IgG ON patients and captures the spectrum of disease presenting to neuro-ophthalmology clinics. Patients were evaluated with a comprehensive neuro-ophthalmic examination that included OCT and formal visual fields. Our study is limited in that it was retrospective in nature and follow-up time was limited, precluding strong conclusions on the effectiveness of long-term immunosuppression. We also may not have captured patients presenting with transverse myelitis or other neurological syndromes as they may have not been routinely referred to neuro-ophthalmology.
In conclusion, this study gives an initial overview of the spectrum of MOG-IgG ON in a group of Canadian patients. Our cohort was different from most previously reported cohorts in the following ways: older onset age; lower incidence of optic disc edema on presentation; lower rates of associated headache and neurological symptoms; less radiologic abnormalities; and lower relapse rates. Despite the poor visual acuity at presentation, most patients recover good visual function.
Acknowledgements
The authors have no financial support or additional contributions to acknowledge.
Conflicts of Interest
None.
Statement of Authorship
Conception and design: JM; Data retrieval: AG, EM, JM; Drafting of manuscript: AG, EM, JM; Critical appraisal: EM, JM; Final approval: EM, JM.



