


Preamble
Concussion, or mild traumatic brain injury (mTBI), has gained significant importance in the literature over the past few years. Although often used as synonyms, mTBI and concussion have slightly varying definitions.Reference Conidi1, Reference Vargas and Dodick2 For the purposes of this paper we will use both terms since they represent clinical practice, as well as keywords in the literature. Mild TBI is defined as a head injury associated with loss of consciousness of <30 minutes, Glasgow Coma Scale of 13 or higher, post-traumatic amnesia <24 hours, altered level of awareness of <24 hours and normal neuroimaging.Reference Pinchefsky, Dubrovsky, Friedman and Shevell3 The most recent consensus statement on concussion in sport defines a concussion as a TBI induced by biomechanical forces and caused by either a direct blow to the head, face, neck or elsewhere on the body with an impulsive force transmitted to the head, which typically results in the rapid onset of short-lived impairment of neurological function that resolves spontaneously, but in some cases, the signs and symptoms evolve over a number of minutes to hours. The acute clinical signs and symptoms reflect a functional disturbance rather than a structural injury and may or may not include loss of consciousness. Resolution of the clinical and cognitive features typically follows a sequential course.Reference McCrory, Meeuwisse and Dvorak4
Sports-related concussions occur between 1.4 and 3.8 million annually in the United States of America, with many going unreported by athletes.Reference Conidi1, Reference Delic, Alhilali, Hughes, Gumus and Fakhran5, Reference Maroon, LePere, Blaylock and Bost6 This number does not include all the non-sports-related concussions or mTBIs. It is estimated that the annual costs related to mTBI in the United States are close to US$17 billion.Reference Delic, Alhilali, Hughes, Gumus and Fakhran5 Thankfully, between 80% and 90% of these patients evolve favourably.Reference Maroon, LePere, Blaylock and Bost6, Reference Ghodadra, Alhilali and Fakhran7 However, given the considerable number of these injuries, a significant minority, between 10% and 20% of these patients, develop chronic symptoms, often referred to as the post-concussive syndrome (PCS).Reference Maroon, LePere, Blaylock and Bost6, Reference Ghodadra, Alhilali and Fakhran7 This syndrome is a constellation of symptoms that ultimately prevent the athlete or patient from successfully returning to play, workplace or school. The most common symptom in PCS is headache,Reference Conidi1, Reference Ghodadra, Alhilali and Fakhran7–Reference Monteith and Borsook10 occurring in up to 88% of sports-related concussions,Reference Conidi1 followed closely and often concomitantly by photophobia. Individually disabling, but collectively worse, these symptoms prove to be a significant challenge to clinicians. The term post-traumatic headache (PTH) will be used in this review to describe headache attributed to traumatic injury to the head which does not include the disorder of headache attributed to whiplash. We reviewed PTH with photophobia together as our clinical experience indicates the combination of these two symptoms was frequent clinically without ever being specifically described in the literature. We hypothesized that there may be an underlying common pathophysiological explanation for this symptom combination.
Objectives
The purpose of this paper is to summarize the current level of knowledge and understanding of the pathophysiological basis behind PTH and photophobia in concussion/mTBI patients. The goal is to guide further fundamental research into the understanding of these mechanisms which will eventually help provide better and more targeted care for affected patients.
Methods
A review of the literature was conducted using the databases CINAHL, EMBASE and PubMed. We have included our search terms and protocol in Annex. All types of articles, including review articles, randomized controlled trials, prospective and retrospective studies, series and case reports, and abstracts were initially included to ensure a global view of the subject. All papers were screened for topics or sections of pathophysiology of PTH or photophobia in patients with mTBI. We included papers published since 2000 in English and in French, and initially included abstracts in order to encompass the largest scope and to minimize publication bias. Owing to the qualitative nature of this review, the goal was to obtain the broadest scope of knowledge about the subject despite some weaker quality papers. We included papers with adult and pediatric populations. We excluded papers specifically addressing moderate to severe TBI but included some papers where there was no distinction between the groups. Through a preliminary screen of titles and abstracts, papers with a potential to discuss the pathophysiology of PTH in mTBI were kept for a full-text screen. Any paper that discussed pathophysiological aspect of PTH was included. For photophobia, the same process was applied but with an even larger scope because of the paucity of literature.
Our original review included 3966 records. Using the database manager EndNote, duplicates were removed, with a resultant 3948 records. An initial screen of titles led to the exclusion of 3548 records. A more in-depth screen of abstracts and full texts, resulted in our final list of records totaling 51 titles which were analyzed and the narrative summary is provided in the following sections. To our knowledge this is the first paper solely focusing on the pathophysiology of PTH and photophobia in concussion/mTBI.
Introduction
Headache is the most common symptom in the acute and chronic phases of any severity of TBI (mild,Reference D’Onofrio, Russo, Conte, Casucci, Tessitore and Tedeschi8, Reference Kjeldgaard, Forchhammer, Teasdale and Jensen9, Reference Blume11–Reference Seifert13 moderate or severe).Reference Defrin, Gruener, Schreiber and Pick14–Reference Mayer, Huber and Peskind16 The majority of these headaches will resolve on their own within 6 months.Reference Erickson, Neely and Theeler12 However, a significant minority of these patients will continue to experience headaches, commonly referred to as PTH. In the United States of America, 1.8 million develop acute PTH and 400,000 people develop chronic PTH annually.Reference Erickson, Neely and Theeler12
Post-traumatic headache are currently classified as a secondary type of headache.Reference Sarmento, Moreira, Brito, Souza, Jevoux and Bigal17 Once the initial diagnosis of PTH is made, the clinical phenotype is then determined based on the more common primary headache types (most commonly tension or migraine). It is unclear whether these phenotypically similar patients have the same underlying pathophysiology or if the TBI “triggers” the same physiological mechanism.Reference D’Onofrio, Russo, Conte, Casucci, Tessitore and Tedeschi8, Reference Mayer, Huber and Peskind16, Reference Heyer and Idris18 Many hypotheses have been discussed, although not exclusively for the mTBI population. Current clinical practice bases its treatment approach for PTH on the clinical phenotype. Most PTH clinically resemble tension or migraine headaches or a mix of the two.Reference D’Onofrio, Russo, Conte, Casucci, Tessitore and Tedeschi8, Reference Daiutolo, Tyburski, Clark and Elliott19 The exact distribution varies, but it is estimated that approximately 33%-50% of PTH have migraine features and 40%-97% have tension-type features.Reference Conidi1, Reference Kjeldgaard, Forchhammer, Teasdale and Jensen9 Once this distinction is made, the clinician treats the patient with the standard therapeutic arsenal for typical tension or migraine headache.
Furthermore, all severities of TBI are often grouped together when discussing PTH and its pathophysiology, although clinical experience and several reports indicate that the mTBI population suffer from more PTH than their moderate-to-severe counterparts.Reference Conidi1, Reference Blume11, Reference Erickson, Neely and Theeler12, Reference Defrin, Riabinin, Feingold, Schreiber and Pick15, Reference Mayer, Huber and Peskind16, Reference Daiutolo, Tyburski, Clark and Elliott19–Reference Riechers, Walker and Ruff24
In the PCS, photophobia is often the second most common symptom after headache and it often accompanies and complicates the headache disorder. This sensitivity to light and visual stimuli, including television or computer screens, is a significant burden for patients. In our clinical experience, it often has a compounding effect with the headache disorder. As with PTH, very little is known about the pathophysiology of photophobia. We explore the literature in an attempt to determine our current level of understanding. We will attempt to link some fundamental science concepts from the review to clinical questions that may help bridge the gap between fundamental understanding and clinical practice.
Post-Traumatic Headaches in mTBI or Concussion
Tension and migraine headaches are discussed separately, although there are probably common mechanisms underlying both phenotypes and that several patients will present a mixed clinical picture in the setting of PTH.
Migraine is a neurovascular condition defined by attacks of moderate to severe throbbing headache. It is often accompanied by sensitivities, notably photophobia and phonophobia, but also sensitivity to odor and movement. Gastrointestinal symptoms are also common, as well as changes in brain states such as emotional, cognitive, autonomic or vestibular.Reference Monteith and Borsook10 Chronic migraine occurs when headaches occur more than 14 days per month for 3 months, where only eight of these headache days need to have migraine characteristics.Reference Cutrer25 Interestingly, head trauma may be a trigger for transformation from episodic to chronic migraine.Reference Garza26
Migraine is believed to be a primary neuronal dysfunction leading to changes, both intracranially and extracranially. The previously popular vascular theory of migraine is no longer considered accurate. The most up-to-date understanding of its pathophysiology relies on a combination of cortical spreading depression, activation of the trigeminovascular system and central and peripheral sensitization. The roles of serotonin and calcitonin gene-related peptide are also appreciated in the pathophysiology of migraines. Cortical spreading depression, a self-propagating wave of neuronal and glial depolarization, is thought to account for the aura of migraine and partially contribute to the pain of migraine through activation of the trigeminovascular system. The sensory neurons originating from the sensory trigeminal ganglion innervate large cerebral vessels, pial vessels, dura mater and large venous sinuses. Once the trigeminal ganglion stimulated, a release of vasoactive neuropeptides occurs and is associated with neurogenic inflammation, which also contributes to the pain component of migraine. Sensitization of these peripheral afferents as well as central sensitization of second order sensory neurons also plays a role in many symptoms associated with migraine.Reference Cutrer25
Tension-type headache (TTH) is the most prevalent headache disorder in the general population, whereas migraine is the most common headache diagnosis in the primary care settingReference Cutrer25, Reference Garza26 as it is generally more severe, and patients with migraine seek medical help. It is characterized by a mild to moderate bilateral headache. It is classically non-throbbing and has no associated symptoms or features. The pathophysiology of TTH is probably multifactorial. A common mechanism to TTH and migraine is the theory of sensitization. Sensitization of peripheral structures is believed to be important in episodic TTH, whereas central sensitization because of prolonged nociceptive input is believed to be the basis for conversion to chronic TTH. It is thought that TTH patients have deficient descending inhibition. There is also some evidence that neurogenic inflammation may also play a role in chronic TTH. Finally, unlike migraine, there seems to be only a minor role of hereditary and genetic factors in episodic TTH to the exception of chronic TTH.Reference Taylor27
Headaches attributed to whiplash and medication overuse headaches are also commonly encountered in the PTH population.Reference Pinchefsky, Dubrovsky, Friedman and Shevell3, Reference Heyer and Idris18 Although clinically important, their pathophysiology will not be reviewed in depth. Whiplash headaches are postulated to be because of concomitant injury to cervical structures. The nociceptive inputs from these structures converge in the trigeminocervical complex and explain the head pain caused by cervical spine injury.Reference Pinchefsky, Dubrovsky, Friedman and Shevell3 Medication overuse headaches is believed to be due to dysregulation of serotonergic and opiate systems and chronic overuse of headache medication, and is mostly associated with opioids and caffeine.Reference Pinchefsky, Dubrovsky, Friedman and Shevell3, Reference Lane and Arciniegas28
A general hypothesis to understanding the pathophysiology of PTH suggests that TBI activates an underlying TTH or migraine headache predisposition and pain then follows, or it accentuates headache patterns that were pre-existent to the trauma.
Acute Versus Persistent PTH
The distinction between acute and persistent PTH is based solely on time since the brain injury, and has a cut-off of 3 months.Reference Bryan and Hernandez29 The pathophysiology of acute and persistent PTH must be viewed as a continuum. Most acute PTH resolves on their own; however, 8%-35% of PTH will chronicize.Reference Erickson, Neely and Theeler12, Reference Bryan and Hernandez29 More emphasis in the literature is placed on persistent PTH, but appreciating the pathophysiology of the acute phase is an important step to understanding the chronic phase. Acute PTH is likely of multifactorial origin. Acutely, PTH patients may have impaired pain inhibition systems and a generalized sensitivity to pain.Reference Lainez, Piera and Bono30 The transition of an acute PTH to persistent PTH is an important clinical and research challenge. The reason for chronicity in some acute PTH remains unclear. Chronic neuroinflammation and concepts of sensitization are possible key mechanisms for persistent PTH.
Pathophysiology
Inflammation
With injury to the central nervous system (CNS), microglia, the main immune cells, become activated and can assume either pro or anti-inflammatory roles. Their anti-inflammatory role involves reparative and regenerative functions important for neuronal survival. The pro-inflammatory response, which includes the release of degradative, toxic and autocrine and paracrine compounds, is intended to clear the CNS of harmful substances, but can be harmful to normal cells. This activation has been histologically described, even in mTBI. Damage to the blood-brain barrier in all severities of TBI can alter its function and permeability, allowing entry of proteins, pathogens and leukocytes, which can all contribute to pro-inflammatory activities and ultimately cell damage. Cytokines which regulate this inflammatory process, normally in low amounts in the CNS, are activated mainly by glial cells and include interleukin-1 and tumor necrosis factor-α. This neuroinflammation, owing to direct injury to trigeminal nerve afferents or structures innervated by the trigeminal nerve, could be a plausible explanation for acute PTH.Reference Mayer, Huber and Peskind16
The role of inflammation in the pathophysiology of PTH is further supported by the findings of pain signaling molecules such as calcitonin gene-related peptide (CGRP) and nitric oxide synthase (NOs) which can activate the trigeminovascular system and trigger migraine headaches.Reference Daiutolo, Tyburski, Clark and Elliott19, Reference Marcus, Altura and Altura21 A mTBI mouse model study was conducted and demonstrated increased levels of these molecules and this increase was associated with trigeminal allodynia as well as photophobia in the acute phase. Increases in CGRP levels were noted at several levels along the trigeminal pathway, even at distant sites from the injury. Notably, meningeal nociceptors were sensitized by this inflammation. Interestingly, treatment with sumatriptan and a CGRP antagonist decreased allodynia and photophobia in this mouse mTBI study. Further supporting the role of an inflammatory process are the findings of increased CGRP and substance P after injury to the somatosensory cortex in mice.Reference Elliott, Oshinsky, Amenta, Awe and Jallo31 The CGRP levels in the brainstem correlated with periorbital allodynia as well.Reference Elliott, Oshinsky, Amenta, Awe and Jallo31 Chronic sensitization in the context of a chronic inflammatory stimulus could be one explanation for the chronic PTH.Reference Mayer, Huber and Peskind16, Reference Daiutolo, Tyburski, Clark and Elliott19, Reference Marcus, Altura and Altura21, Reference Newberg, Serruya and Gepty32 However, this theory alone would in difficult explain the higher incidence of PTH in mTBI as the more severe TBI would likely be more inflammatory.
The role of inflammation in the pain-sensitive dura has been implicated. The concept of sterile meningeal inflammation induced particularly by dural mast cells was demonstrated in an animal model of mTBI. Acute mast cell degranulation ipsilateral to the side of the mTBI was found up to 30 days and is thought to be due to the mechanical transmission of forces to the dura. Interestingly, mast cell degranulation was also noted on the contralateral side.Reference Levy, Edut and Baraz-Goldstein33 Could persistence of this dural inflammation lead to sensitization and ultimately contribute to persistent PTH?
An interesting study on systemic inflammation and mTBI using high-sensitivity C-reactive protein (HS-CRP) levels demonstrated correlation between elevated HS-CRP and unfavorable outcomes in this population during the first 3 months post-mTBI. Higher CRP levels were associated with persistent PCSs, psychological problems and cognitive impairment. It did not correlate with physiological symptoms such as headache, but it should be noted that headache is a symptom used in the diagnosis of PCS.Reference Su, Xu and Li34
Peripheral Origin
A model for mTBI on mice study showed that in the acute phase post-mTBI, there was selective enhancement of nociceptive responses to cranial structures (specifically the calvarial periosteum—considered a deep cranial structure by the authors) but not to extra-cranial structures (specifically the hindpaw). Moreover, there is evidence of inflammatory changes in deep cranial structures (mast cell degranulation) suggesting that in the acute phase post-mTBI, the PTH may originate from peripheral rather than from central or supraspinal pain pathways.Reference Conidi1 It is important to distinguish these findings which apply to acute PTH and may contrast with theories of central sensitization explaining how PTH can become chronic.Reference Daiutolo, Tyburski, Clark and Elliott19, Reference Marcus, Altura and Altura21 It can be therefore questioned if more aggressive treatment of PTH in the acute phase could prevent this central sensitization phenomenonReference Elliott, Oshinsky, Amenta, Awe and Jallo31 and thus prevent persistent PTH from developing.
A peripheral component to PTH can also be suspected based on findings that PTH patients also display cranial mechanical hyperalgesia, probably because of injury to peripheral structures, including blood vessels, nerve fibers and bone, as well as the initiation of an inflammatory cascade. This process may once again possibly contribute to sensitization.Reference Defrin, Gruener, Schreiber and Pick14, Reference Defrin, Riabinin, Feingold, Schreiber and Pick15
Central Origin
Structural brain matter damage has been described even in mTBI. It has been postulated that damage anywhere along the pain pathways can be a source of central pain.Reference Defrin, Gruener, Schreiber and Pick14, Reference Marcus, Altura and Altura21 Damage to spinothalamic or thalamocortical pathways would be most implicated and may occur even with mTBI.Reference Defrin, Riabinin, Feingold, Schreiber and Pick15 Patients with PTH tend to have increased heat-pain thresholds which is in favor of a central origin.Reference Defrin, Gruener, Schreiber and Pick14 Loss or a decrease of descending pain inhibition and loss of opioidergic systems could also contribute to PTH, a mechanism also reported in TTH and migraine.Reference Defrin, Gruener, Schreiber and Pick14, Reference Defrin, Riabinin, Feingold, Schreiber and Pick15, Reference Marcus, Altura and Altura21
Further support for a central origin comes from imaging studies on mTBI patients with PTH migraines. Advanced magnetic resonance with diffusion-tensor imaging and proton spectroscopy is being increasingly studied as a means for the diagnosis of concussion or mTBI and PCS.Reference Ghodadra, Alhilali and Fakhran7, Reference Mayer, Huber and Peskind16 These imaging techniques were shown to differentiate mTBI patients from controls, as well as distinguish mTBI patients with migraines from mTBI patients without migraine based on white matter injuries.Reference Delic, Alhilali, Hughes, Gumus and Fakhran5 The imaging techniques seem to be able to detect white matter injury patterns that correlate with PTH.Reference Ghodadra, Alhilali and Fakhran7 The role of these white matter changes in the pathogenesis of PTH is less clear, however.Reference Ghodadra, Alhilali and Fakhran7, Reference Torrente, Cabezas, Avila, Garcia-Segura, Barreto and Guedes35
Functional rather than structural brain abnormalities are becoming increasingly recognized in the mTBI population as standard imaging modalities that often demonstrate no abnormalities. Blood flow abnormalities and damage to neurotransmitter systems (such as dopamine) post TBI are being evaluated with single-photon emission computed tomography. Mild TBI patients with PTH have been shown to have lower cerebral blood flow in the right frontal lobe and increased cerebral blood flow in the left parietal lobe when compared to controls.Reference Newberg, Serruya and Gepty32 These findings may be because of diffuse axonal injury and inflammation;Reference Newberg, Serruya and Gepty32 however, causality has yet to be determined.
Cortical Spreading Depolarization
Cortical spreading depolarization (CSD) is a pathological depolarizing phenomenon that has been described in several pathological states such as strokes, epilepsy, intracranial hemorrhages and migraines and TBI. It interrupts local cortical function for minutes to hours.Reference Torrente, Cabezas, Avila, Garcia-Segura, Barreto and Guedes35 Glutamate-induced toxicity seems to be the common triggering mechanism for CSD, which ultimately leads to a depression in cortical activity. Concomitantly, there is also a pathological change in vascular response, where vasoconstriction, instead of vasodilation, occurs, which further contributes to brain injury.Reference Kramer, Fujii, Ohiorhenuan and Liu36 Traumatic brain injury-induced hypermetabolism in conjunction with reduced blood flow leads to a metabolic mismatch.Reference Seifert and Shipman37 Traumatic brain injury promotes an environment susceptible to CSD,Reference Seifert13 and patients exhibiting CSD tend to have worse outcome;Reference Kramer, Fujii, Ohiorhenuan and Liu36 however, it is unclear how this applies to the mTBI population. The link between CSD and PTH is largely through the findings of CSD in the pathophysiology of migraines.Reference Monteith and Borsook10 Injury-triggered CSD is more commonly seen with severe TBI, however.Reference Monteith and Borsook10
Glutamate
It is known that TBI results in glutamate release proportional to the extent of the TBI. Glutamate release from astrocytes and microglia occurs after a TBI. This effect of excess glutamate is calcium influx and neuronal injury.Reference Maroon, LePere, Blaylock and Bost6, Reference Packard38 Furthermore, the excess glutamate overstimulates receptors, notably NMDA receptors, and creates ion imbalances.Reference Packard38 The effect of this excitotoxicity on the pathogenesis of PTH is unclear but can be due to its ability to promote CSD and central sensitization.Reference Riechers, Walker and Ruff24, Reference Packard38
Autoimmune
One study found that auto-antibodies to glutamate receptors in pediatric persistent PTH were elevated even in the mild closed TBI. These auto-antibodies would be the result of hypoxic and neurometabolic changes in the brain.Reference Goryunova, Bazarnaya and Sorokina39 There is a net result of glutamate receptor overstimulation.Reference Goryunova, Bazarnaya and Sorokina39
Electrolyte abnormalities
Magnesium is required to regulate transmembrane electrical activity and has a neuroprotective action by decreasing the excitotoxic effects of glutamate and N-methyl-D-aspartate (NMDA).Reference Maroon, LePere, Blaylock and Bost6 A pediatric study demonstrated that PTH patients had lower levels of ionized magnesium and higher ratios of ionized calcium to ionized magnesium. It is unclear if there is a causal relationship between these electrolytes findings and the pathophysiology of PTH.Reference Marcus, Altura and Altura21
Genetic
A genetic predisposition to symptoms after mTBI has been postulated after reports of severe neurological symptoms in mTBI patients who are carriers of a CACNA1A gene mutation, an ion channelopathy associated with familial hemiplegic migraine.Reference Seifert13 This theory supports the idea by which some patients would present a genetically lower threshold to symptom development after a head traumaReference Seifert13 and could help explain the discrepancy in symptom severity after a similar intensity trauma. This specific genetic mutation has not been directly linked to PTH; however, it does provide insight toward a genetic avenue in understanding the pathophysiology of PTH.
Hormonal Dysregulation
Hormonal dysregulation has been observed in TBI patients, including hypothalamic dysfunction, which could mediate headaches and be a potential contributor to PTH.Reference Monteith and Borsook10 Hypothalamic injury in TBI may contribute to PTH through its effect on the sleep–wake cycle and circadian rhythms.Reference Monteith and Borsook10 It is important to note that this finding is not specific nor associated with mTBI and is usually seen in more severe TBI. Therefore, its role in PTH in the mTBI population remains speculative.
Clinical Risk Factors
Different risk factors for the development of PTH have been identified with epidemiological studies. Women tend to develop more PTH. The mechanism of injury also seems to have an impact as motor vehicle accidents tend to predispose more to PTH. Pre-trauma history of headache, lower education and socioeconomic levels, short duration of post-traumatic amnesia and medication overuse are other risk factors for the development for PTH.Reference D’Onofrio, Russo, Conte, Casucci, Tessitore and Tedeschi8, Reference Erickson, Neely and Theeler12 As with many pain conditions, comorbid psychiatric conditions such as anxiety or post-traumatic stress disorder (PTSD), and ongoing financial or legal proceedings regarding the injury can cloud the picture.Reference D’Onofrio, Russo, Conte, Casucci, Tessitore and Tedeschi8, Reference Erickson, Neely and Theeler12, Reference Defrin, Gruener, Schreiber and Pick14, Reference Defrin, Riabinin, Feingold, Schreiber and Pick15, Reference Heyer and Idris18, Reference Marcus, Altura and Altura21
Photophobia in mTBI or Concussion
Photophobia, or photosensitivity, is defined as an abnormal tolerance to light or visual discomfort in the presence of normal light levels.Reference Digre and Brennan40–Reference Truong, Ciuffreda, Han and Suchoff42 Although occasionally reported in isolation, photophobia is commonly seen in patients with PTH and commonly associated with migraine-type headaches.Reference Katz and Digre41 Tension-type headache sufferers also report more sensitivity to light compared with controls.Reference Digre and Brennan40
Photophobia is an increased sensitivity to light and visual stimuli from all sources. Patients tend to be particularly sensitive to artificial indoor light, computer monitors including screens of all kind and glare.Reference Katz and Digre41 Screen intolerance is particularly important given its omnipresence and patients will often dim the light intensity of the screen or modify the hue.Reference Clark, Hasselfeld, Bigsby and Divine43 This may be due to the rate at which computer or cell phone screens refresh (60 Hz) with concussed individuals having a higher critical flicker frequency threshold.Reference Mansur, Hauer and Hussain44 Of particular interest, migraineurs report a high prevalence of photophobia during a headache attack, some report light as a trigger and some report light sensitivity even outside of their headaches.Reference Digre and Brennan40, Reference Katz and Digre41
Photophobia is a common and debilitating symptom in mTBI patients and PCS,Reference Digre and Brennan40, Reference Katz and Digre41, Reference Bulson, Jun and Hayes45, Reference Goodrich, Flyg, Kirby, Chang and Martinsen46 and even more so when associated with PTH. A military study found a 22% prevalence of photophobia in troops with mTBI.Reference Digre and Brennan40 It may have a significant impact on the patient’s quality of life.
It is not uncommon for patients with photophobia to be wearing sunglasses in a physician’s waiting room.Reference Katz and Digre41 In fact, colored glasses have been studied as a treatment method for patients suffering from light sensitivity. Blue, green, red and purple-colored glasses seemed to offer the most relief from light sensitivity. This therapeutic option can be helpful in alleviating symptoms of light sensitivity post-mTBI and may help with the recovery process.Reference Blumenfeld47
Clinical experience has shown that subtle photophobia in mTBI patients may be underappreciated clinically and can be discovered with proper patient questioning. Part of the cause for underrecognizing this symptom may be because of the lack of standardized tests for the diagnosis or quantification of this symptom.Reference Truong, Ciuffreda, Han and Suchoff42
Photophobia is probably a response to light to protect the retina, but in some this threshold is lower than normal.Reference Katz and Digre41, Reference Mansur, Hauer and Hussain44 Retinal ganglion cells (RGC) are the cells which send axons to make up the optic nerve and collect information from photoreceptors.Reference Blumenfeld47 Retinal ganglion cells send their axons to the lateral geniculate nucleus.Reference Blumenfeld47 Intrinsically photosensitive RGC, also known as melanopsin cells, have been discovered and are postulated as the key cell in photophobia. These cells contain the photopigment melanopsin, which are activated via traditional visual pathways (photoreceptors such as rods and cones) or can be activated by light directly. These cells make up to <1% of ganglion cells.Reference Katz and Digre41
There are currently three postulated pathways involved in photophobia. The first pathway begins with “regular” ganglion cells which project via the optic nerve to the olivary pretectal nucleus, then to the superior salivatory nucleus. Through the pterygopalatine ganglion, this causes ocular vasodilation and activation of ocular trigeminal afferents through the trigemino-automic relfex.Reference Digre and Brennan40 These afferents then project to the trigeminal nucleus caudalis, the thalamus and to the cortex.Reference Digre and Brennan40, Reference Katz and Digre41 The trigeminal system is key in the pathophysiology of photophobia as it is very closely linked to pain sensation.Reference Digre and Brennan40 The ophthalmic division of the trigeminal nerve (V1) is responsible for nociceptive afferents from the eye and orbit.Reference Digre and Brennan40
The second pathway involves the previously described intrinsically photosensitive RGC. These cells, also through the optic nerve, project directly to the thalamus which is also receiving intracranial nociceptive input. The thalamus receives light and pain information, which is then projected to sensory and association cortices. The pathway involving the thalamus is particularly interesting because of the role of the thalamus for multiple sensory integration.Reference Digre and Brennan40, Reference Katz and Digre41 This pathway could explain the photophobia seen in patients with meningitis or sub-arachnoid hemorrhage.Reference Digre and Brennan40
The third pathway also involves intrinsically photosensitive RGC, but through projections that do not involve the optic nerve. These cells have been found in rodent iris and could activate trigeminal ocular afferents. This pathway could explain direct trigeminal stimulation in the absence of a functioning optic nerve,Reference Digre and Brennan40, Reference Katz and Digre41 and could help explain photophobia seen in some blind patients.Reference Digre and Brennan40
These pathways are supported by findings on functional MRI which show increased activity in patients with photophobia in the trigeminal nucleus, superior colliculus, thalamus and cortex.Reference Katz and Digre41
An interesting report of three patients who had complete lack of photophobia because of bilateral ventral occipital lobe lesions from cerebral infarction points to another potentially crucial cortical area of the brain needed for photphobia,Reference Mansur, Hauer and Hussain44 which may compliment the above described pathways. This region of the brain, however, is not a classic site of injury in the context of TBI.
CGRP and NO are important inflammatory pain signaling molecules that can activate the trigeminovascular system and are believed to be part of the pathophysiology of PTH as described earlier.Reference Daiutolo, Tyburski, Clark and Elliott19 Blocking CGRP in a mouse TBI model was found to relieve photosensitivity.Reference Daiutolo, Tyburski, Clark and Elliott19
If photophobia persists in the chronic phase, it can often be attributed to concomitant migraine, as photophobia is a key diagnostic element,Reference Digre and Brennan40 and potentially even if the PTH phenotype is of the tension type.
Cortical damage has also been postulated to explain photophobia in TBI. Traumatic brain injury patients with photosensitivity presented with elevated dark adaptation thresholds, which are usually associated with retinal dysfunction. However, the subjects had no retinal dysfunction, which prompted the hypothesis of either post-retinal, cortical or subcortical processes to explain the abnormal dark adaptation with normal retinal function. Head trauma induces changes that result in a hyperexcitable state and increased neural gain which would lead to photosensitivity. A cortical adaptive mechanism attempts to attenuate light sensitivity at all levels that could translate into elevated dark adaptation thresholds.Reference Du, Ciuffreda and Kapoor48 The exact sites involved in the initial injury and postulated adaptive mechanisms require further research. The concept of cortical excitability has also been reported in conditions associated with photophobia, such as migraine and epilepsy.Reference Magone, Kwon and Shin49
As with PTH, a biopsychosocial view of the subject is necessary. Viewing photophobia as a chronic “pain” condition may be necessary to understand the impact of comorbid anxiety, depression or PTSD. A study has showed more light sensitivity in TBI patients with PTSD than in TBI patients without PTSD.Reference Goodrich, Martinsen, Flyg, Kirby, Garvert and Tyler50
Discussion
Post-traumatic headache and photophobia in the mTBI population are challenging symptoms for clinicians to treat. In order to provide better clinical care for these patients, more fundamental research is needed to help understand the underlying mechanisms behind the symptoms. Although it is often reported that little is known about the pathophysiology, this review highlights significant research advances.
It is likely that mechanisms of PTH overlap with primary TTH and migraine, but trauma does in fact cloud the clinical and pathophysiological picture. Post-traumatic headache is likely a multifactorial process that evolves over time from the acute to the chronic phase. The final pain-producing step is probably common to the traumatic and atrauamtic headache types, as evidenced by the same clinical phenotypes. However, the steps or mechanisms by which trauma ends up triggering these similar mechanisms are crucial to uncover.
We propose the following diagram (Figure 1), adapted from Defrin et al,Reference Defrin51 to describe our summarized understanding of the pathophysiological mechanisms to PTH and photophobia in mTBI as well as possible overlapping pathophysiological mechanisms between the two symptoms.
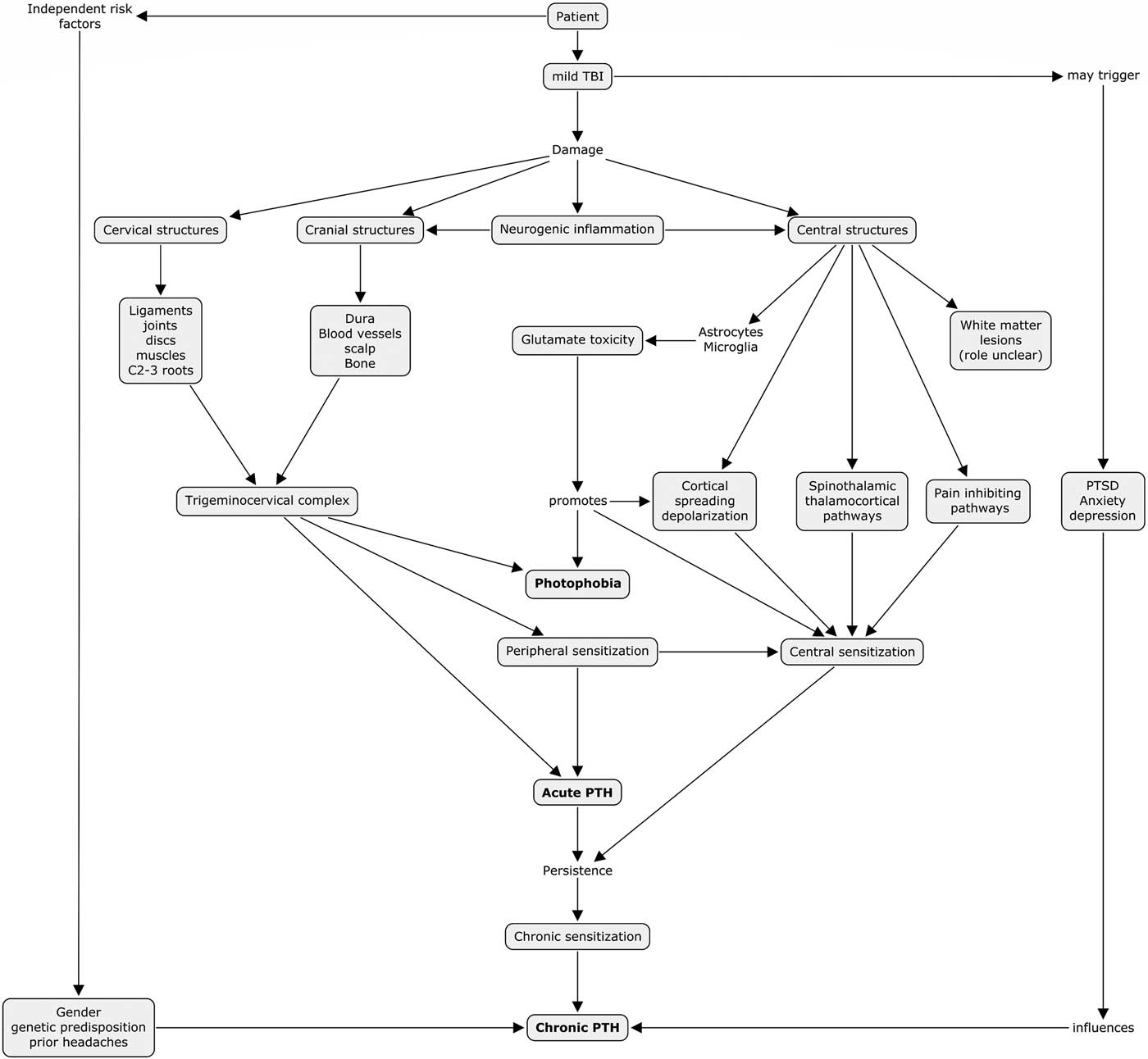
Figure 1 Proposed pathophysiological mechanisms to PTH and photophobia in mTBI. PTH=post-traumatic headache, mTBI=mild traumatic brain injury, PTSD=post-traumatic stress disorder.
A multifactorial explanation to PTH and photophobia is evident and it is unlikely that a single eventual explanation exists. Anatomically, interactions between injured cervical, cranial and central neurological structures are evident and their respective contributions probably vary between individual patients. We highlight an interesting finding from our narrative review, which shows overlapping mechanisms between PTH and photophobia in the mTBI population through both peripheral and central mechanisms. This pathophysiological finding correlates with our clinical experience where patients with PTH often experience concomitant photophobia. Furthermore, it is not uncommon that photophobia is the more functionally limiting symptom as it limits going outdoors, interacting in visually stimulating environments and using computers or other electronic devices. Targeting these common mechanisms could lead to a breakthrough common treatment for both symptoms.
In order to precisely describe the mechanisms, we believe that focussing on mTBI and separating it from the moderate to severe counterparts is crucial. Further distinguishing the subpopulation of sports-related concussions may also help as our experience shows that this population is quite different in the clinical setting. Furthermore, although overlap and mixed features probably exist, it could be beneficial to study mTBI PTH by dividing them into groups based on their clinical phenotypes (tension vs. migraine).
Our understanding of the pathophysiology of photophobia in general has also progressed. There is still more work to be done in order to determine how we can apply this knowledge clinically. Overlapping pathophysiological data is documented between PTH and photophobia, notably, the implication of the trigeminovascular system, dural irritation and inflammation, cortical hyperexcitability and migraines. A common pathway for both PTH and photophobia would be pleasing to the medical field, but more research is needed to prove this.
Some questions do remain. Why do patients with mTBI suffer from more PTH? Why do some patients develop a migraine PTH versus a tension type PTH? Can aggressive treatment of acute PTH prevent sensitization and the development of persistent PTH?
Conclusion
In conclusion, we believe that this narrative review highlights the current level of understanding of the pathophysiology of PTH and photophobia in patients with mTBI. These patients may be seen by a variety of specialists, including primary care, sports medicine, neurology, neurosurgery and physical medicine, and thus it is important that fundamental and clinical evidence be made available for all clinicians. We hope this review can be used as a guide for future fundamental and clinical research.
Annex
Search terms and protocol
photophobia[mh] OR headache[mh] OR migraine disorders[mh] OR headache disorders[mh] OR photophobia*[ot] OR headache*[ot] OR migraine*[ot] OR head pain*[ot] OR cranial pain*[ot] OR cephalalgia*[ot] OR cephalodynia*[ot] OR hemicrania*[ot] OR light sensitivit*[ot] OR photophobia*[tiab] OR headache*[tiab] OR migraine*[tiab] OR head pain*[tiab]OR cranial pain*[tiab] OR cephalalgia*[tiab] OR cephalodynia*[tiab] OR hemicrania*[tiab] OR light sensitivit*[tiab]
AND
rain concussion[mesh:noexp] OR post-concussion syndrome[mh] OR çraniocerebral trauma[mesh:noexp] OR “head Injuries, closed“[mesh:noexp] OR brain injuries[mesh:noexp] OR (athletic injuries[mh] AND (brain[tiab] OR head[tiab] OR skull[tiab])) OR brain concussion*[ot] OR commotiocerebri[ot] OR post concussion syndrome[ot] OR post concussive syndrome[ot] OR craniocerebral trauma*[ot] OR craniocerebral injur*[ot] OR “parietal trauma*”[ot] OR crushing skull injur*[ot] OR “occipital trauma*”[ot] OR “temporal trauma*”[ot]OR “frontal trauma*”[ot] OR “forehead trauma*”[ot] OR head injur*[ot] OR head trauma*[ot] OR brain injur*[ot] OR brain trauma*[ot] OR TBI[ot] OR TBIs[ot] OR brain damage*[ot]OR (athletic injuries[ot] AND (brain[ot] OR head[ot] OR skull[ot])) OR brain concussion*[tiab] OR commotiocerebri[tiab] OR post concussion syndrome[tiab] OR post concussive syndrome[tiab] OR craniocerebral trauma*[tiab] OR craniocerebral injur*[tiab] OR “parietal trauma*”[tiab] OR crushing skull injur*[tiab] OR “occipital trauma*”[tiab] OR “temporal trauma*”[tiab] OR “frontal trauma*”[tiab] OR “forehead trauma*”[tiab] OR head injur*[tiab] OR head trauma*[tiab] OR brain injur*[tiab] OR brain trauma*[tiab] OR TBI[tiab] ORTBIs[tiab] OR brain damage*[tiab] OR (athletic injuries[tiab] AND (brain[tiab] OR head[tiab] OR skull[tiab]))

