Malignant optic glioma of adulthood (MOGA) has a poor prognosis and proclivity to mimic other diagnoses. We describe a middle-aged man with presumed high-grade MOGA of the optic nerve and chiasm who, after failing radiotherapy treatment alone, responded well to three cycles of temozolomide, followed by lomustine. He re-presented two years later with extensive cervical cord lesion leading to eventual demise one month later. This case demonstrates rapid progression of MOGA from involving optic nerve alone to chiasm, emphasizes its malignant behavior, including its potential for non-contiguous metastasis along the neuro-axis, and describes the most effective algorithm to date in treating it.
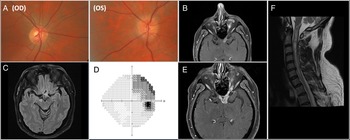
A 47-year-old man noticed rapidly progressing blurry vision in the left eye (LE). He did not seek care for six weeks. On neuro-ophthalmic consultation seven weeks after symptoms onset, vision was 20/20 in the right eye (RE) and light perception in LE, with brisk left relative afferent pupillary defect and elevated optic disc in LE (Fig. 1a). Visual field was normal in RE but demonstrated diffuse depression in LE. Atypical retrobulbar optic neuritis was suspected. MRI brain and orbits revealed enlarged and enhancing left pre-chiasmatic optic nerve, with enhancement/enlargement extending to the left side of optic chiasm (Fig. 1b). There was also a non-enhancing hyperintense lesion in the right cerebral peduncle on T2/fluid attenuation inversion recovery sequence (Fig. 1c). Demyelination was deemed the most likely culprit. Myelin oligodendrocyte glycoprotein (MOG) and neuromyelitis optica (NMO) antibody titers were negative. Lumbar puncture demonstrated normal cerebrospinal fluid (CSF) composition, normal cytology, and negative oligoclonal bands. Computed tomography of the body was unremarkable.
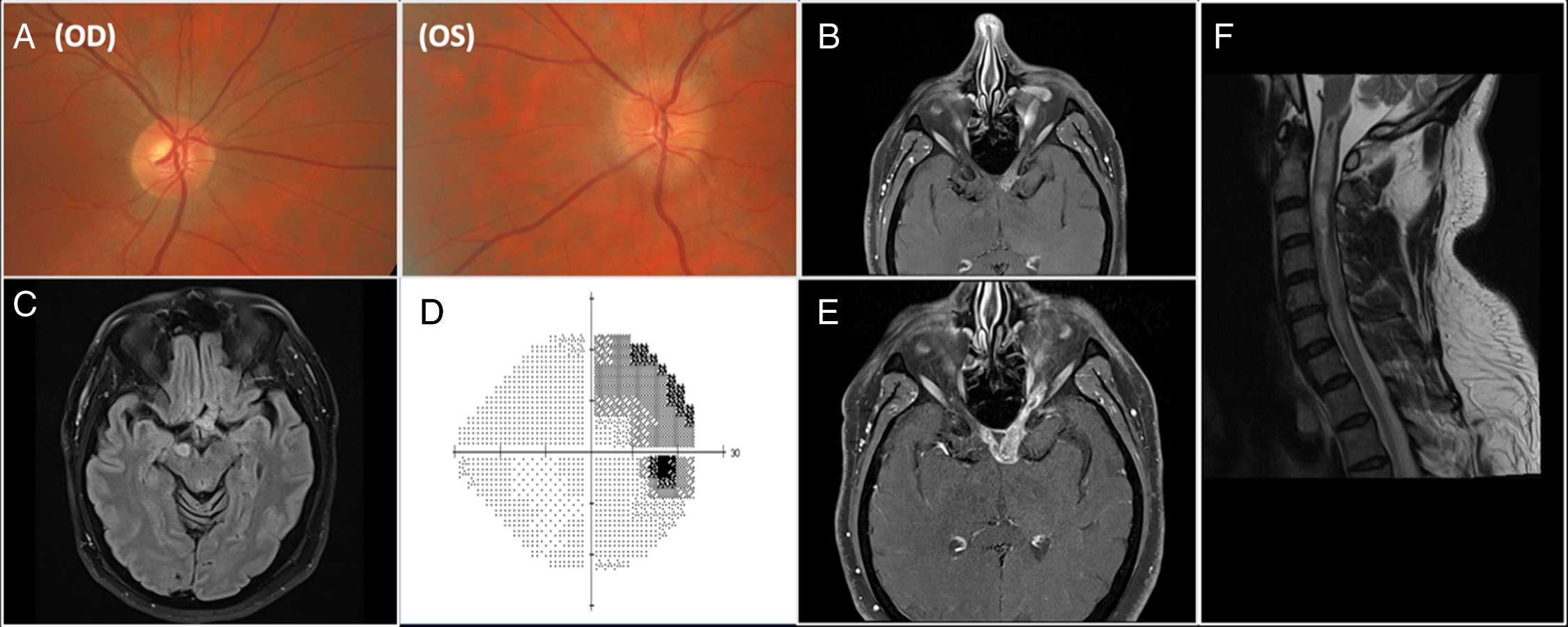
Figure 1: a. Color photographs of optic discs demonstrating normal disc on the right and diffusely elevated optic disc on the left with blurry margins. 1b. Axial T1 post-contrast MR sequence showing diffusely thickened and enhancing left intracranial optic nerve with abnormal signal extending to the optic chiasm. 1c. Axial fluid attenuation inversion recovery MRI sequence showing a right cerebral peduncle lesion. 1d. Visual field (Humphrey 24-2 algorithm) in the right eye demonstrating superotemporal defect respecting vertical midline. 1e. Axial T1 post-contrast MR sequence showing further enhancement and longitudinally extensive enlargement of the left intracranial optic nerve, extending now to the right side of the optic chiasm. 1 f. Sagittal T2 post-contrast MR sequence of the cervical cord showing extensive hyperintensity extending from cervico-medullary junction to T2 level.
Treatment with intravenous methylprednisolone (1 g for 5 days) followed by 60 mg of oral prednisone commenced, however, when vision did not improve after one week, and therapy with seven cycles of plasma exchange was started. One month later, the patient noticed blurry vision in RE. Visual acuity was 20/25 in RE, and no light perception in LE and visual fields demonstrated a superotemporal defect in RE that respected vertical meridian (Fig. 1d), consistent with junctional scotoma. MRI showed greater enlargement of left pre-chiasmatic optic nerve with extension of the lesion to the right side of chiasm (Fig. 1e). Malignant optic nerve/chiasmal glioma was felt to be the most likely diagnosis because of the rapid progression of radiological abnormalities and no response to treatment with corticosteroids. Biopsy of the lesion was offered but repeatedly declined by the patient. Treatment with fractionated radiotherapy (60 Gy in 30 fractions) for presumed MOGA commenced.
One month later, the examination was unchanged. Treatment with three cycles of temozolomide commenced. Six months after initial presentation, there was further enlargement of left pre-chiasmatic optic nerve and optic chiasm on MRI. The lesion in the cerebral peduncle demonstrated minimal enlargement and enhancement, which was attributed to either post-radiation changes or disease progression. Given these findings, chemotherapy was switched to six cycles of lomustine. Eighteen months after initial presentation, there was decrease in optic nerve and chiasmal enlargement on MRI.
Patient remained in extended remission for two years when he re-presented with sudden a onset of right-sided weakness. Examination confirmed severe weakness of the right upper and lower extremities, with decreased pain and temperature sensation along the left C4-level sensory region. Brain MRI was unchanged but spinal MRI demonstrated extensive elongated and expansile central intramedullary hyperintensity that extended from cervico-medullary junction to mid-T2 level with minimal diffusion restriction and patchy enhancement (Fig. 1f). MOG and NMO antibody titers were negative again and CSF contents were normal. Multi-disciplinary tumor board concluded that spinal cord lesion represented a spread of MOGA. The patient subsequently experienced a rapid worsening of respiratory status requiring intubation. The family declined further care and he passed away six weeks later.
With only over 70 reported cases, MOGA is a rare diagnosis that mimics demyelinating optic neuritis and NMO/MOG-related optic neuritis and chiasmitis. Reference Alabiad, Shah, Eatz, Sternau and Lam1,Reference Huang, Patel and Patel2 It should be suspected in all patients with presumed optic neuritis who demonstrate no visual improvement after treatment with corticosteroids and show further optic nerve and/or chiasm enlargement on follow-up neuroimaging with otherwise negative work-up for inflammatory and infiltrative causes of optic neuropathy. The definitive diagnosis requires biopsy, however, characteristic clinical presentation and otherwise negative work-up may obviate the need for biopsy. Reference Huang, Patel and Patel2
MOGA’s presentation contrasts with indolent low-grade optic glioma of childhood, which is often associated with neurofibromatosis type 1 and carries minimal malignant potential. Reference Huang, Patel and Patel2 MOGA typically demonstrates aggressive behavior with tumor infiltration and compression of surrounding brain structures. Patients present with acute vision loss, that over 5-6 weeks progresses to bilateral complete vision loss in 63%. Reference Huang, Patel and Patel2 Neuroimaging typically demonstrates fusiform enlargement and enhancement of the intracranial optic nerve and chiasm. Reference Huang, Patel and Patel2 In 50% of cases, retrograde tumor progression extends to the hypothalamus, basal ganglia, and temporal lobe. Reference Huang, Patel and Patel2 Rarely, MOGA may lead to distal spinal metastasis. Reference Murphy, Timms, McKelvie, Dowling and Trost3
Our case demonstrated distal MOGA spread along neuro-axis without radiological evidence of direct extension: an intramedullary lesion extending down to thoracic cord was presumed to represent distal metastases of the optic pathway lesion with the spinal seeding likely occurring via CSF spread. Reference Mariniello, Peca and Del Basso De Caro4 This is similar to other malignant brain gliomas which typically infiltrate surrounding brain tissues and spread along white matter tracts. Reference Murphy, Timms, McKelvie, Dowling and Trost3
Patients with MOGA have a median life expectancy of less than one year with statistically significant improvement in survival in those who receive combination chemo- and radiotherapy over radiotherapy alone. Reference Alireza, Amelot, Chauvet, Terrier, Lot and Bekaert5 Adjunct therapy with lomustine after disease recurrence is another option to extend survival. Reference Huang, Patel and Patel2 There are only a few rare reports in the literature of patients with the long survival (4.5–7 years). Reference Alabiad, Shah, Eatz, Sternau and Lam1 Compared to the median life expectancy of MOGA, our patient had extended survival on a regimen of temozolomide and radiotherapy, surviving two years after presumptive diagnosis before expiry. Nonetheless, determination of prognosis and the extent of our patient’s benefit on this regimen was limited by their refusal of biopsy and subsequent pathologic confirmation with molecular classification. Indeed, while there is a dearth of molecularly characterized optic gliomas in the literature, molecular variants may have unique mutations that confer variable response to therapy and represent an opportunity for optimized molecular-targeted treatment. Reference Samples, Mulcahy Levy and Hankinson6 Such investigation was precluded in our patient, though their initial symptomatic presentation was markedly controlled with sustained remission for two years on their treatment regimen.
As in our case, patients may be reluctant to undergo a biopsy for suspected MOGA, especially given the irreversibility of visual loss after biopsy. However, forgoing biopsy and molecular characterization may preclude analysis of prognosis and determining sensitivity to various treatment regimens. Reference Samples, Mulcahy Levy and Hankinson6 This implies an urgent need for non-invasive diagnostic techniques, for which various alternatives are emerging. Promising non-invasive diagnostic technologies include liquid biopsy, involving the detection of tumor markers in body fluids (e.g., blood, CSF, or urine), and radiomics, which correlates molecular and subcellular tumor features with imaging heterogeneity. Reference Bonosi, Ferini and Giammalva7,Reference Gerardi, Cannella and Bonosi8 Such techniques, if eventually clinically validated, standardized, and available at large-scale, may increase rates of elective biopsy and offer clinicians fulsome guidance to clinical management with minimized patient risk.
In summary, we described a case of presumed MOGA that presented with optic nerve involvement but quickly spread to involve the chiasm. After extended remission, patient re-presented two years later with distal non-contiguous metastasis. This case reminds neuro-ophthalmologists that MOGA should be suspected in all patients with presumed atypical optic neuritis that does not respond to corticosteroids and demonstrates further enlargement/enhancement of affected portion of visual pathway on imaging. It also describes the most effective treatment strategy to date in managing this ultimately fatal disease.
Acknowledgements
None.
Statement of authorship
All authors contributed equally to data gathering, preparation, manuscript writing, revision drafting, and final approval of the manuscript.
Financial support
None.
Competing interests
None.
Statement of ethics
This study was granted approval from the University of Toronto Health Sciences Research Ethics Board. This work uses anonymized data and will not generate new forms of identifiable information.