



Introduction
Predominantly sleep-related hypermotor seizures frequently originate from the frontal lobe (referred to as nocturnal frontal lobe epilepsy [NFLE]). Because it is increasingly recognized that such seizures may sometimes arise from extra-frontal regions, a consensus panel recently recommended replacing the term NFLE to sleep-related hypermotor epilepsy (SHE). Reference Tinuper, Bisulli and Cross1 Some patients show an autosomal dominant mode of inheritance (ADNFLE). To date, pathogenic variants in six genes have been associated with ADNFLE: CHRNA4 and CHRNB2 (coding, respectively, for the α4 and β2 subunits of the neuronal nicotinic acetylcholine receptor –nAChR), CHRNA2, CRH, KCNT1, and DEPDC5. Reference Nobili, Proserpio and Combi2 We report here two cases of insular epilepsy, one with a variant in CHRNB2 and the other in CHRNA4.
Methods
Patients
The patients affected were evaluated for predominantly sleep-related hypermotor seizures with a semiology suggesting an extra-frontal onset. Each individual underwent advanced epileptological work-up. The investigations included familial history enquiries, neuropsychological and neurological examination, scalp video-electroencephalography (EEG) monitoring, neuroimaging (both morphological and functional), and intracranial study (one case). For the first patient, CHRNB2 (NM_000748), CHRNA2 (NM_000742), and CHRNA4 (NM_000744) were sequenced by Sanger, and CHRNA4 was analyzed for deletion/duplication using quantitative polymerase chain reaction (qPCR). For the second patient, a panel of 36 genes associated with epilepsy was sequenced using next-generation sequencing (Supplementary Table 1).
Cell Culture and Transfection Procedure
Either wild-type (WT) or mutant β2 (NM_000748) constructs were transiently cotransfected with a vector expressing the α4 (NM_000744) subunit in human embryonic kidney (HEK) cells (TsA201 subclone; American Type Culture Collection, Manassas, VA, USA) using standard procedures. Reference Conti, Aracri and Chiti3 Briefly, cells were cultured in Dulbecco’s Modified Eagle Medium-high glucose (DMEM to high glucose) supplemented with 10% fetal calf serum (Sigma-Aldrich, St. Louis, MO, USA) and 2 mM L-glutamine, at 37°C and 5% CO2. Cells were seeded in 35-mm Petri dishes and transfected with Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA). Constructs ratios were 1:1 for α4:β2 (WT), 1:1 for α4:β2-Thr26Met (homozygous mutants), and 2:1:1 for α4:β2-Thr26Met:β2 (simulated heterozygotes). The DNA concentration in the transfection mixture was 4 µg of nAChR subunit constructs plus 0.6 µg of Enhanced Green Fluorescent Protein (E-GFP) pcDNA3 (Clontech Laboratories, Mountain View, CA, USA). E-GFP expression allowed easier detection of transfected cells. Cells were incubated with the transfection mixture for 4 to 6 h. Next, the receptors’ expression was increased by transferring the transfected cells to lower temperature (29°C–30°C, in 5% CO2), 24 h before patch-clamp recording. Reference Cooper, Harkness, Baker and Millar4
Patch-Clamp Experiments
Cells were voltage-clamped in the whole-cell configuration, 36 to 72 h after transfection, with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA), at room temperature (22°C to 24°C). Micropipettes (2–3 MΩ) were pulled from borosilicate capillaries with a P-97 Flaming/Brown Micropipette Puller (Sutter Instrument Co., Novato, CA, USA). Cell capacitance and series resistance were compensated (up to approximately 75%). Fluorescent cells were identified with an Eclipse TE200 microscope (Nikon Instruments, Sesto Fiorentino, Italy) equipped with a fluorescence-integrated transmission electron microscopy epifluorescence attachment. Currents were low-pass-filtered at 2 kHz and acquired online at 5 to 10 kHz with pClamp 9.2 hardware and software (Molecular Devices). Drugs were applied with an RSC-160 Rapid Solution Changer (Bio-Logic Science Instruments, Claix, France).
Solutions and Drugs
Unless otherwise specified, chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). The extracellular solution contained (in mM): NaCl 130, KCl 5, CaCl2 2, MgCl2 2, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) 10, and D-glucose 5 (pH 7.3). Patch pipettes contained (in mM): K-gluconate 140, KCl 5, MgCl2 1, 1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) 0.5, HEPES-KOH 10, Mg-adenosine 5′-triphosphate 2, and Na-guanosine 5′-triphosphate 0.3 (pH 7.3). Stock solutions of nicotine (10 mM) were prepared weekly in our extracellular solution and kept refrigerated. Extracellular solutions with the appropriate nicotine concentrations were prepared daily; pH was always readjusted after nicotine addition.
Analysis of Patch-Clamp Data
Data were analyzed with Clampfit 9.2 (Molecular Devices) and OriginPro 9.1 (OriginLab, Northampton, MA, USA). To measure the reversal potential (V rev) of WT and mutant currents, current–voltage (I/V) relations were obtained by applying 1 s voltage ramps from −60 to +10 mV, in the absence or presence of 100 µM nicotine. To isolate the nicotinic currents, the background currents were always subtracted to those obtained in the presence of the agonist. To decrease the fluctuations in V rev measurements, we generally averaged the currents obtained from at least three consecutive voltage ramps. In the presence of nicotine, ramps were applied during the slow phase of current desensitization. The effects of channel desensitization are irrelevant for V rev estimates. No correction of liquid-junction potential was applied to any of the voltage values given in the text.
mRNA Quantification
Total RNA was isolated from cultured cells using Direct-zolTM RNA MiniPrep (Zymo Research) and eluted in water. 1 µg of the total extracted amount of RNA was subsequently treated with DNase I and reverse-transcribed using SuperScriptTM VILOTM cDNA Synthesis Kit (Invitrogen). The first-strand cDNA was used as a template for real-time polymerase chain reaction (PCR) using a human CHRNB2-specific primer pair (forward [FW] 5′-GGAGCAGATCATGACCACCA-3′ and reverse [RV] 5′-TGCCGTGGTCTCCTATGATG-3′) and EvaGreen fluorescent dye (Biorad). β-actin was used as housekeeping gene (FW 5′-CGACAGGATGCAGAAGGAG-3′ and RV 5′-ACATCTGCTGGAAGGTGGA-3′). PCR reaction was performed using a CFX96 real-time system (Bio-Rad, Hercules, CA, USA) sequence detector. Data, normalized for β-actin gene, are expressed as fold change value respect to the untransfected cells according to the 2-[ΔΔC(t)] algorithm.
Statistical Analysis
Unless otherwise indicated, data are given as mean values ± SEM. For patch-clamp data, “n” represents the number of tested cells. Statistical comparisons between two populations of data were carried out with Student’s t-test for unpaired samples, after checking for data normality (with Kolmogorov–Smirnov test) and variance homogeneity (with F-test). In case of variance heterogeneity, the Welch’s correction was applied. In case of non-normal data, we reported the median values and used the Mann–Whitney test to assess statistical significance.
Data Availability
Unpublished anonymized clinical data and experimental data are available upon request from the corresponding author.
Results
The first patient is an ambidextrous girl with mild language delay, now 14 years old, who started having seizures at the age of 4 years. The paroxysmal events were characterized by an unpleasant tingling sensation in the lower back, right arm, and both legs, followed by fear and complex motor behaviors for ~30 seconds. Seizures became predominantly nocturnal after a few weeks, recurring in clusters every two days. After failing six antiepileptic drugs, the patient was referred for epilepsy surgery (at 9 years old). Investigations revealed right centro-temporal spikes on scalp EEG, normal brain magnetic resonance imaging (MRI) (1.5 and 3 Tesla), and right insular activation on ictal single photon emission computed tomography (Figure 1). Source localization of interictal and ictal discharges recorded during magnetoencephalography (CTF 275-sensor system, Philips Medical Systems, Best, the Netherlands; Canada) using an electrical current dipole model revealed a tight cluster at the posterior end of the right Sylvian fissure (posterior insula > parietal operculum > temporal operculum). Resection of the right posterior insula and overlying parietal and temporal opercula resulted in seizure freedom except for three possible auras of tingling sensations in the back for a few seconds (Engel 1b; follow-up 4 years). Pathological evaluation of the resected tissue was not possible because of the technique of subpial aspiration of the cortex. Gene dosage analysis by qPCR did not reveal deletion/duplication within or including the CHRNA4 gene. Sanger sequencing of CHRNB2 revealed a heterozygous missense variant (c.77C>T; p. Thr26Met) in the proband and her asymptomatic father. This variant is found in 0.1% of European non-Finnish alleles of the gnomAD dataset (136/126556 alleles) Reference Lek, Karczewski and Minikel5 and is predicted to be deleterious by sorting intolerant from tolerant (SIFT), Reference Kumar, Henikoff and Ng6 and probably damaging by PolyPhen2. Reference Adzhubei, Schmidt and Peshkin7 It is also classified as likely benign by two independent clinical laboratories in the ClinVar repository.

Figure 1: Epileptic focus localization and surgery of patient 1. (A) Surface EEG recorded active interictal epileptiform discharges over the right centro-temporal regions [filters HFF (high filter frequency) = 35 Hz, LFF (low filter frequency) = 1 Hz; 30 mm/s; 10 µV/mm]. (B & C) Ictal single photon emission computed tomography coronal and axial images demonstrated right operculoinsular hyperperfusion. (D & E) Source localization of interictal and ictal epileptiform discharges overlaid onto coronal and axial MRI images. One electroclinical seizure was captured during magnetoencephalography with onset of repetitive discharges over the right central, parietal, and temporal channels. Ictal (red) and interictal (yellow) dipole clusters localized over the posterior third of the right Sylvian fissure (posterior insula and parietal operculum more than temporal operculum). (F) Surgical resection of the parietal more than temporal operculum and subsequent subpial removal of the posterior insula.
We tested the effect of this variant by co-expressing the β2 WT subunit and/or β2-Thr26Met with α4, in HEK TsA201 cells. Figure 2A and 2B shows representative whole-cell current traces elicited by the indicated concentration of nicotine, at −70 mV, in cells, respectively, expressing WT or mutant homozygous nAChRs, as indicated. The maximal current was tested at regular intervals to rule out nAChR rundown, which is sometimes observed in cell lines. Nicotine was preferred to the physiologic agonist acetylcholine, to avoid applying muscarinic receptor blockers, whose effect on different nAChR subtypes is not well defined. Reference Zwart and Vijverberg8 The concentration-response relations generated from these experiments are shown in Figure 2C, which plots the peak whole-cell currents normalized to the current measured at 100 µM nicotine, for WT, homozygotes, and simulated heterozygotes. Figure 3A shows the corresponding non-normalized average whole-cell current densities (i.e., for each cell, the peak current was divided by the cell capacitance). Overall, minor differences were observed between the shapes of the concentration-response relations obtained from cells expressing WT or mutant nAChRs subunits. Moreover, no significant difference between WT and heterozygous receptors was observed in the kinetics of channel desensitization and deactivation (data not shown). These results suggest that β2-Thr267Met did not produce major alterations in the receptor’s kinetics or sensitivity to the agonist. To assess whether the mutant subunit altered ion selectivity, we measured the current reversal potential (V rev) from current/voltage (I/V) relations obtained by applying voltage ramps from −60 to +10 mV. Representative I/V curves from cells expressing WT or heterozygous nAChRs are shown in Figure 3B. On average, V rev was −4.56 ± 1.28 mV for WT receptors (n = 6), −2.79 ± 1.74 mV in the homozygous mutant form (n = 7; p > 0.05 compared to WT, with t-test), and −2.9 ± 2.6 mV in the simulated heterozygote (n = 5; p > 0.05, compared to WT). These results suggest that β2-Thr26Met does not produce major alterations in channel permeability, which is consistent with the location of Thr26, considerably distant from the conduction pore.
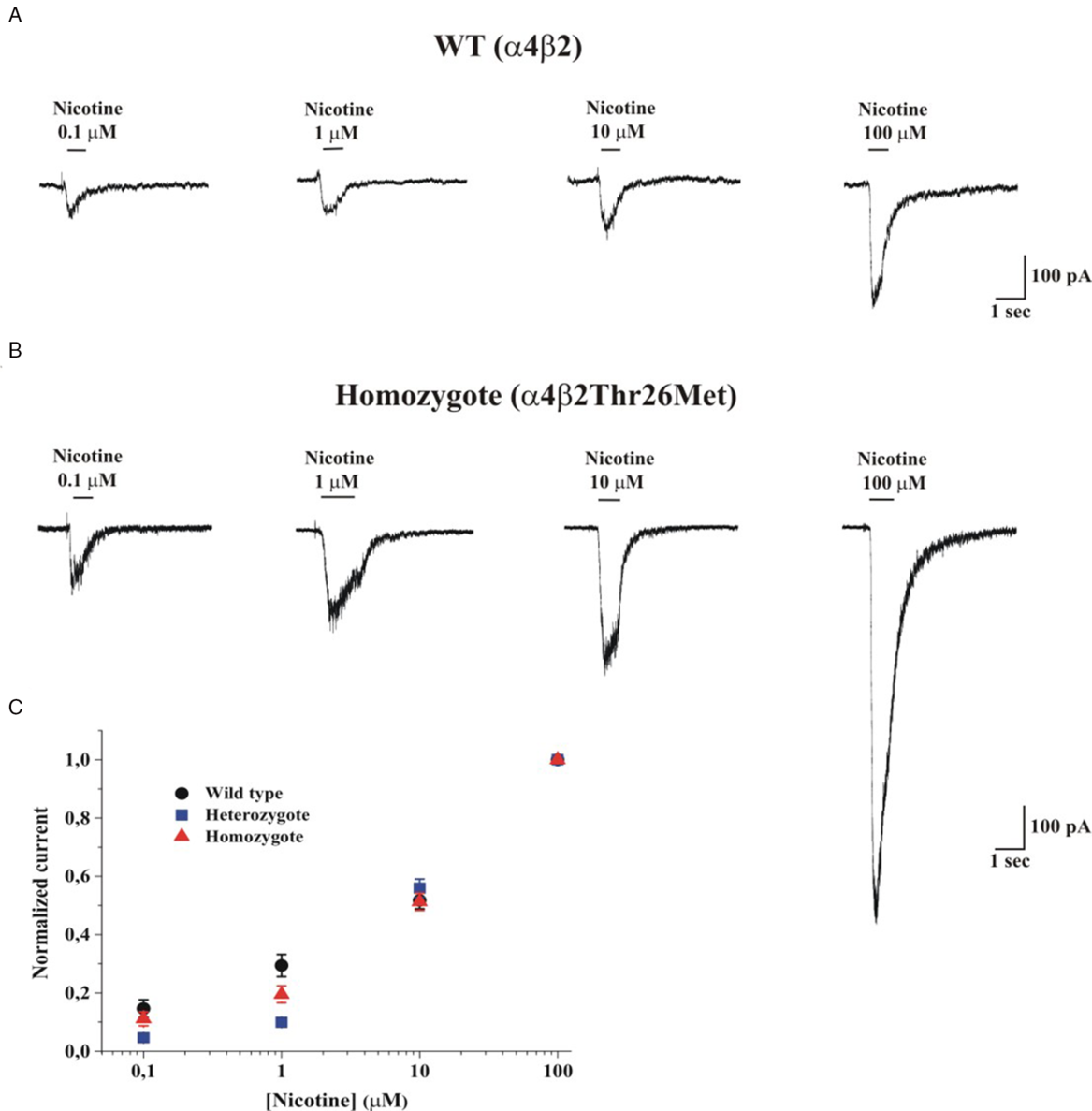
Figure 2: Whole-cell current traces elicited by nicotine at −70 mV. (A) Representative whole-cell current traces elicited by the indicated concentration of nicotine in HEK cells expressing a4β2 (WT) nAChRs; V m was −70 mV. (B) Same as A, for mutant α4β2Thr26Met (Homozygote) nAChRs. The continuous lines above the current traces mark the time of nicotine application. (C) Concentration-response relations obtained by applying different nicotine concentrations to WT (circles), heterozygous (squares), and homozygous (triangles) receptors. The applied nicotine concentrations were 0.1, 1, 10, and 100 µM. V m was −70 mV. Data points are average peak currents normalized to the maximal value and plotted as a function of ligand concentration. Data summarize the results obtained from 65 cells, in 10 runs of transfection.

Figure 3: Comparison of elicited currents in wild, homozygote, and heterozygote types. (A) Average peak current densities, for the indicated nAChR types, at increasing concentrations of nicotine (0.1, 1, 10, and 100 µM). Currents were measured as illustrated in Fig. 2 and divided for the cell capacitance. The statistical comparison between the currents obtained at 100 µM nicotine is given in the main text. (B) I/V relationships for WT and heterozygous receptors, as indicated. Currents were elicited by applying voltage ramps from −60 to +10 mV (duration was 1 s). The current flowing through nAChRs was isolated by subtracting the background current from the current recorded in the presence of 100 µM nicotine. The illustrated currents are averages of three trials, applied consecutively (interval between trials was 1 s). V rev was generally between −10 and 0 mV. Full statistics are given in the main text. (C) The distribution of individual whole-cell current amplitudes elicited by 100 µM nicotine is shown for cells expressing WT (black circles) and homozygous (red triangles) receptors. The superimposed Tukey box plots include the central 50% of data points, with horizontal lines denoting median values. Whiskers’ length is 1.5 times the interquartile range. Open squares denote mean values. (D) Same as C, but showing the distribution of current densities (pA/pF).
As indicated by the representative current traces shown in Figure 2 as well as the average current densities (Figure 3A), the main effect produced by β2-Thr26Met was a considerable increase in the whole-cell current amplitude, which was approximately tripled compared to the WT. Figure 3C shows the peak whole-cell current values in cells expressing WT (n = 26) or mutant homozygotes (n = 24), and treated with 100 µM nicotine. The median values were, respectively, 177 and 515 pA (p = 0.005, with Mann–Whitney test). Figure 3C shows the current densities calculated from the same experiments, in which the source of variability caused by the different cell dimensions is minimized. The median current densities were 2.54 for WT (n = 26), 6.09 for the homozygotes (n = 24; p = 0.003 compared to WT, with Mann–Whitney test), and 5.87 for the heterozygotes (n = 15; p = 0.0008 compared to WT). Further statistics are illustrated in the figure panels. We conclude that the presence of β2-Thr26Met markedly increases the functional nAChR expression. Thus, we performed reverse transcription-PCR assays on the transfected cultures to check the relative expression of CHRNB2 in the presence or absence of the variant (Figure 4). These experiments showed no significant differences between cultures transfected with vectors containing the cDNA carrying WT or the mutant sequence, indicating that no major alterations are produced by the mutant gene at the transcriptional level. Further work will be required to assess whether the effect of β2-Thr26Met is caused by a higher nAChR expression onto the plasma membrane, or an increase in the channel open probability at a given concentration of agonist, or a combination of these factors.

Figure 4: β2 nAChRs subunit levels of expression detected by real-time quantitative PCR in HEK cells transfected with either WT or mutant β2 (p. Thr26Met) constructs. Data represent the mean ± SEM and are expressed as fold increase of mRNA levels normalized to a housekeeping control gene (β-actin) and to non-transfected HEK cells (NT). No significant differences in β2 transcription levels were observed between the WT and mutant β2 constructs (214.31 ± 87.10 versus 271.08 ± 63.95, p > 0.050 with unpaired t-test; n = 3).
While the study was ongoing, we identified a second case of insular epilepsy associated with a variant affecting a subunit of the nAChR. The patient, a 34-year-old right-handed woman with seizures since the age of 5 months, was referred to our institution for epilepsy surgery evaluation after failing 10 antiepileptic drug trials. Seizures were predominantly sleep-related, recurring daily. Seizures recorded during video-EEG monitoring were characterized by an initial arousal, anxiety, auditory disturbances (“sounds were duller”), and a sensation of body warmth followed (after a mean latency of 15 s) by axial and pelvic movements, vocalization, a frightened facial expression, and then an asymmetric tonic or dystonic posture. Rarely, seizures could evolve into bilateral tonic–clonic movements. EEG recordings revealed bilateral independent temporo–frontal spikes, electrical seizures involving independently left or right temporal leads, and electroclinical seizures with diffuse onset. Magnetoencephalographic spike source localization (CTF 275-sensor system, Canada) using an electrical current dipole model revealed clusters over both insulae extending to the posterior orbito–frontal regions (Figure 5). Brain MRI was normal twice (1.5 and 3 Tesla), and positron emission tomography (PET) showed a mild left insulo-temporal hypometabolism. Genetic testing disclosed a heterozygous missense variant in CHRNA4 (c.1079G>A; p. Arg360Gln). This variant is absent from population databases Reference Lek, Karczewski and Minikel5 and is predicted to be deleterious and probably damaging by SIFT and PolyPhen2. Reference Kumar, Henikoff and Ng6,Reference Adzhubei, Schmidt and Peshkin7 This variant was not previously reported in patients. Her estranged brother who also had epilepsy was contacted but declined to share clinical information and undergo genetic testing. Fenofibrate failed to improve seizure frequency, while nicotine patches (7 mg/day) managed to transiently reduce seizure frequency by approximately 20–30%. Reference Puligheddu, Melis and Pillolla9,Reference Willoughby, Pope and Eaton10 The patient was also heterozygous for a variant in PRICKLE1 (NM_153026.2) (c.151T>C; p. Cys51Arg). This variant was found at the highest frequency in Latino alleles from the Broad gnomAD dataset (0.0087%, 3/34572). It is predicted to be deleterious and benign by SIFT and PolyPhen2, respectively, Reference Lek, Karczewski and Minikel5–Reference Adzhubei, Schmidt and Peshkin7 and it has not been reported in ClinVar.

Figure 5: Epileptic focus localization of patient 2. (A) Surface EEG recorded frequent spikes over the left fronto–parieto–temporal (A1 and A2) and right fronto–temporal leads (A2) [filters HFF (high filter frequency) = 70 Hz, LFF (low filter frequency) = 1 Hz; 30 mm/s; 7, 5 µV/mm]. (B) Source localization of interictal epileptiform discharges overlaid onto axial (B1), coronal (B2), and sagittal (B3). MRI images showing dipole clusters localized over the right (upper panel) and left (lower panel) insulo-orbitofrontal regions.
Discussion
In this report, we present two patients with sleep-related insular epilepsy harboring missense variants in genes coding for nAChR subunits. To our knowledge, such an association has not been previously reported. The literature does, however, provide supporting evidence linking ADNFLE/SHE, the insula, and nAChRs. First, it has been demonstrated in genetic studies and animal models of seizures that nAChR activity is altered in ADNFLE/SHE. Reference Ghasemi and Hadipour-Niktarash11–Reference Nichols, Henderson and Marotta14 Moreover, a few authors have reported a significant antiseizure effect of transdermal nicotine patches in ADNFLE/SHE cases involving CHRNA4 Reference Lossius, de Saint Martin and Myren-Svelstad15,Reference Pavlakis and Douglass16 or CHRNB2 Reference Sieciechowicz and Kohrman17 mutations. This effect, partially observed in case 2, has mainly been attributed to a desensitization of nAChR resulting from therapeutic excessive exposure to nicotine. Reference Pavlakis and Douglass16,Reference Sieciechowicz and Kohrman17 Second, it has been shown that SHE may occasionally originate from extra-frontal regions, notably the insulo-opercular region. Reference Ryvlin, Minotti and Demarquay18–Reference Proserpio, Cossu and Francione21 Indeed, we found at least 16 cases of nocturnal seizures of operculo-insular origin in the literature. Reference Ryvlin, Minotti and Demarquay18-Reference Proserpio, Cossu and Francione21 Four of these patients were reported to have a family history of seizures suggesting a strong genetic component, albeit genetic testing was performed (or mentioned) for only one with no evidence of CHRNA4 and CHRNB2 variants. Reference Ryvlin, Minotti and Demarquay18–Reference Proserpio, Cossu and Francione21 Third, a study using 2-[18F] F-A-85380 PET has shown that cortical nAChR density was highest in the insular and anterior cingulate cortices. Reference Picard, Sadaghiani, Leroy, Courvoisier, Maroy and Bottlaender22 This observation could explain why some patients with CHRNA4 and CHRNB2 variants have seizures starting preferentially in those locations. Fourth, tractography data in humans have confirmed a wide network of connections between the insula and the ventro-mesial and dorso-lateral prefrontal cortex, providing a structural network for insular seizures to propagate and generate classical sleep-related hypermotor seizures. Reference Ghaziri, Tucholka and Girard23
Based on tools predicting the possible impact of amino acid substitutions on the structure and function of proteins (SIFT and PolyPhen-2), variants identified in this report could be deleterious. In addition, we performed functional characterization of the p. Thr26Met missense variant in HEK cells. The main alteration produced by β2Thr26Met was an increase in whole-cell current density, which was not accompanied by major alterations in channel permeability. These results suggest that the average number of open channels at a given concentration of agonist is higher in the presence of β2Thr26Met. However, because of the well-known difficulties of carrying out extensive single-channel analysis with nAChRs, we cannot presently distinguish whether the observed effect is caused by an increased amount of nAChR protein in the plasma membrane, or a higher probability that a channel is open, or both. Regardless of the specific mechanism, our results are broadly consistent with the observation that mutant α4 and β2 nAChR subunits linked to ADNFLE/SHE often confer gain-of-function properties to α4β2 nAChRs in different expression systems. Reference Zwart and Vijverberg8,Reference Willoughby, Pope and Eaton10,Reference Ghasemi and Hadipour-Niktarash11,Reference Picard, Sadaghiani, Leroy, Courvoisier, Maroy and Bottlaender22 The second patient carried a p. Arg360Gln (c.1079G>A) missense variant in CHRNA4 absent from population databases and predicted to be damaging. Although we cannot rule out a possible contribution of the missense variant in PRICKLE1 to the patient’s epileptic condition, we feel it is unlikely since this gene has mainly been associated with progressive myoclonic epilepsy and autism spectrum disorder (EPM1A, OMIM: 612437).
Data from population databases have shown that CHRNA4 and CHRNB2 are constrained for missense variants suggesting that such variants are more likely to be associated with disease. Reference MacArthur, Manolio and Dimmock24 This is consistent with the literature as functional evidence for pathogenicity has been provided for missense variants in CHRNB2 and CHRNA4 found in families with epilepsy (NFLE). Reference Becchetti, Aracri, Meneghini, Brusco and Amadeo25 As it was for our first patient, unaffected carriers were also previously identified indicating that other factors might also be implicated in the pathogenesis in those patients. This is consistent with the rare variant hypothesis supporting the notion that complex diseases such as epilepsy could be explained by a number of genetic variants of low effect on the phenotype. Reference Chen, Wang and Wang26,Reference Schork, Murray, Frazer and Topol27 At the moment, the evidence available to clinically assess the pathogenicity of the p. Thr26Met (c.77C>T) missense variant in CHRNB2 is conflicting or insufficient. Although it has been suggested that variants found in more than one heterozygote in population databases are less likely to be pathogenic, Reference Bennett, Petrovski, Oliver and Berkovic28 we provide functional evidence that indicates that it could play a role in the disease. Also, it is important to note that Clinvar submitters mostly rely on existing literature to assess variant pathogenicity. Therefore, their interpretation of the p. Thr26Met (c.77C>T) missense variant could change over time, as more data become available. In the case of the p. Arg360Gln missense variant in CHRNA4, there are no experimental evidence demonstrating its impact on the protein, but it is absent from population databases and predicted to be damaging by in sillico analysis. Therefore, using the American College of Medical Genetics and Genomics guidelines for interpretation of sequence variants, we classify the variants reported in this study as variants of unknown significance. It is important to note that those guidelines were designed for the evaluation of patients with primarily Mendelian disorder and might not be suited for complex diseases such as epilepsy. Reference Richards, Aziz and Bale29 Since genomewide screening for pathogenic point mutations or structural variants was not performed, we cannot rule out the presence of other deleterious genetic alteration and that the variants reported here are rare benign polymorphisms.
We can also hypothesize that for our patients, two variants in the same gene, one affecting the germline and one somatic affecting brain cells, are required for the expression of the epileptic phenotype. Therefore, both alleles of the same gene have to carry a genetic variant for the disease to manifest. This “two-hit” mutational mechanism was originally formulated by Knudson in 1971 for oncogenesis Reference Knudson30 and has since been adapted for various diseases, including epilepsy. Reference Ishida, Picard and Rudolf31–Reference Baulac34 More specifically, it has been described in various lesional and nonlesional epileptic disorders such as familial focal epilepsy with variable foci or ADNFLE. Reference Ishida, Picard and Rudolf31–Reference Iffland, Carson, Bordey and Crino36 The “two-hit” mutational hypothesis may explain the presence of the c.77C>T (p. Thr26Met) variant in CHRNB2 in the asymptomatic father of one of our patients. While both this patient and her father are carrying the c.77C>T (p. Thr26Met) germline variant in CHRNB2, only she would by carrying the second, somatic, CHRNB2 variant in her brain cells resulting in the epileptogenesis. Alternatively, in both patients, it is possible that a localized insult to the brain acquired postnatally combined with the identified germline variants incites epileptogenesis. Overall, we can hypothesize that our reported germline variants may predispose patients to epilepsy, and in concert with a second factor (e.g., somatic variant, brain injury, or other acquired change) would lead to full disease expression.
Conclusions
Overall, our results suggest that CHRNB2 and CHRNA4 variants may be involved in the pathogenesis of not only ADNFLE but also insular epilepsy. The functional study of β2-Thr26Met suggests that the pathogenetic mechanisms may be similar. We hope this paper will entice other groups to report additional similar cases which could further support the association between these genetic variants and nonlesional insular epilepsies.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Acknowledgments
The authors would like to thank the Genome Aggregation Database (gnomAD) and the groups that provided exome and genome variant data to this resource. A full list of contributing groups can be found at http://gnomad.broadinstitute.org/about.
Disclosure
The authors report no disclosure.
Statement of authorship
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/cjn.2020.126.







