

A 25-year-old man noticed gradual worsening of vision in the left eye over 4 weeks. Vision was 20/40 with the presence of optic nerve head swelling on the affected side. Brain MRI reported white matter hyperintense lesions limited to the left frontal lobe. Lumbar puncture demonstrated normal cerebrospinal fluid composition and no oligoclonal banding. Multiple sclerosis was felt to be a possible diagnosis and treatment with dimethyl-fumarate commenced.
Vision continued to deteriorate and when seen 2 months later, it was 20/200 in the left eye with the presence of brisk relative afferent pupillary defect. Fundoscopy demonstrated massive optic nerve head edema with surrounding hard exudates (Figure 1A). Urgent brain and orbits MRI/MRV demonstrated hyperintense white matter lesions in the deep left frontal lobe (Figure 1B) that seemed microischemic and not demyelinating in nature but was otherwise normal. Treatment with dimethyl-fumarate was stopped. Extensive workup (testing for HIV, Lyme, Toxoplasma, Bartonella titers, VDRL, ACE, ANA, ds-DNA, ENA, NMO, total body CT, and gallium scan) looking for potential etiologies of severe unilateral optic nerve head swelling was all unrevealing. Over the following 2 years, vision remained stable with remarkably persistent swelling of the left optic nerve head. Repeated tests including repeat of previously performed serologic testing, total body CT, and lumbar puncture were all noncontributory.
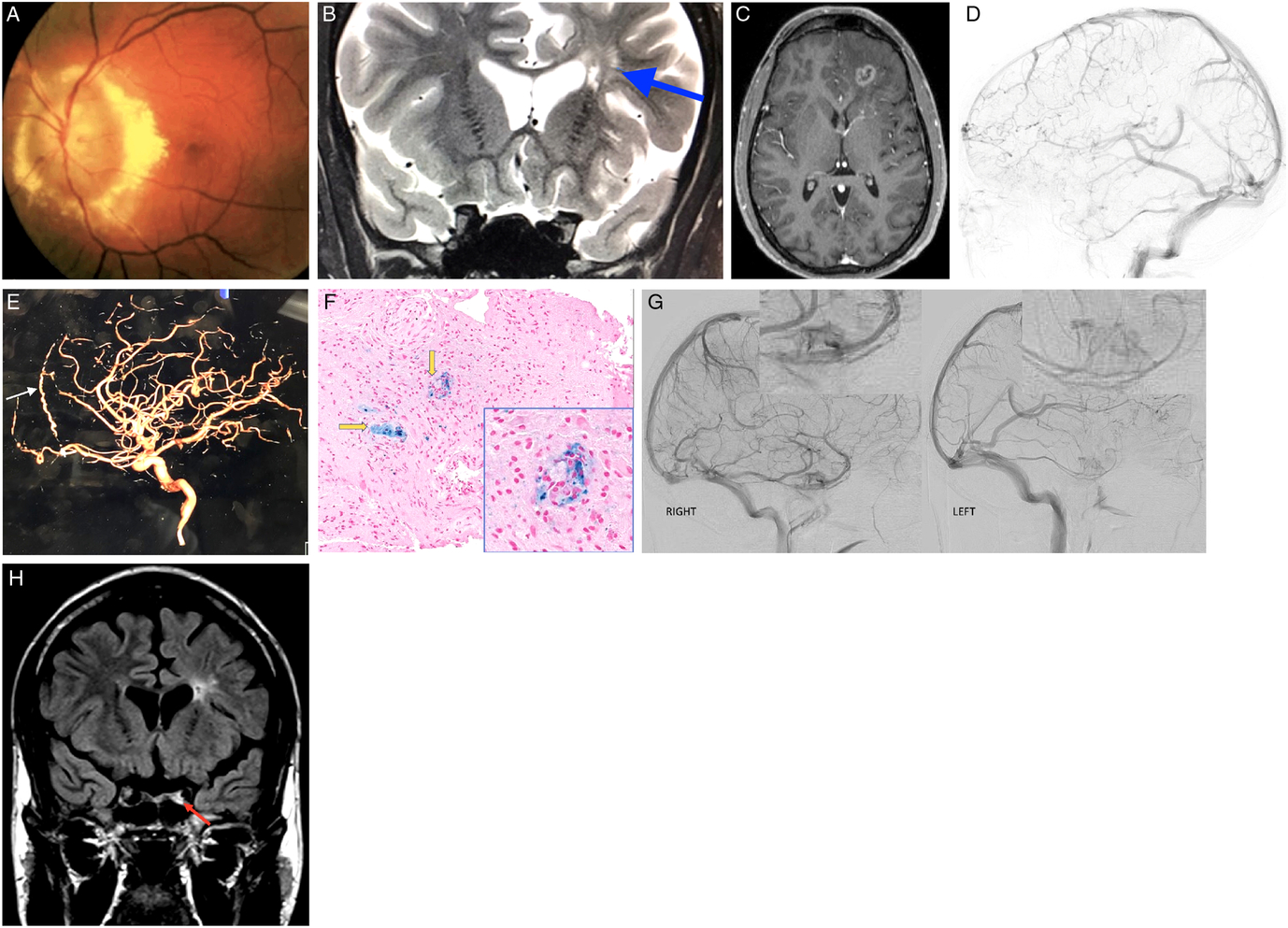
Figure 1: (A) Fundus photo of the left eye demonstrating massive optic nerve head swelling with surrounding hard exudates. (B) MRI. Corontal T2 sequence demonstrating hyperintense lesions deep in the left frontal lobe (blue arrow). (C) MRI. Axial T1 with gadolinium demonstrating left frontal mass lesion with peripheral enhancement. (D) Left lateral internal carotid angiogram demonstrating "pseudophlebitic pattern" in the left frontal lobe. (E) Left lateral internal carotid angiogram demonstrating enlarged left anterior falcine artery (white arrow). (F) Frontal lobe biopsy specimen. Hematoxylin and eosin (H&E) and Prussian blue stain demonstrating perivascular macrophages (yellow arrows). (G) Right and left lateral carotid angiograms demonstrating decreased left cavernous perfusion. (H) MRI. Coronal FLAIR demonstrating left hyperintense signal in the left cavernous sinus corresponding to cavernous sinus thrombosis.
Two and a half years after the initial presentation, the patient developed a generalized seizure. MRI of the brain was urgently performed and demonstrated a left frontal lobe mass with surrounding edema and peripheral enhancement (Figure 1C). Brain biopsy was performed and demonstrated macrophages surrounding blood vessels and unexpected areas of necrosis but was deemed indeterminate. Second opinion on the results of the biopsy reported findings inconsistent with both demyelination and arterial infarct. Cerebral angiography was performed and demonstrated unusual left hemispheric cortical veins without arteriovenous shunting. After consulting with pulmonary, rheumatological, and neuroradiological services, neurosarcoidosis was felt to be a unifying diagnosis. After receiving 3 days of 1 g of intravenous methylprednisolone, treatment with oral mycophenolate mofetil commenced. MRI of the brain performed 2 months later demonstrated dramatic resolution of left frontal lobe lesion.
While satisfied with radiological improvement, the case was reexamined and the diagnosis of neurosarcoidosis was challenged. Cerebral angiogram was meticulously reexamined. Unusual tortuous cortical veins in the left hemisphere were noticed and felt to be compatible with “pseudophlebitic” pattern seen in brain dural arteriovenous fistulas (bdAVF) (Figure 1D).Reference Geibprasert, Pongpech, Jiarakongmun, Shroff, Armstrong and Krings1 Enlarged left anterior falcine artery was also demonstrated (Figure 1E). Review of the literature on the pathological findings seen in bdAVFs yielded one published retrospective study of spinal dural AVFs biopsies that were performed because the lesion resembled a neoplasm radiologically. Their reported tissue findings were the presence of thick-hyalinized vessels in all cases, hemosiderin-laden macrophages in 71%, and necrosis in 29%.Reference Rodriguez, Crum, Krauss, Scheithauer and Giannini2 Third review of brain biopsy was thus requested and reported the presence of thick-hyalinized vessels, hemosiderin-laden macrophages, and areas of necrosis in our case that were all compatible with chronic venous congestion (Figure 1F).
A decision was made to perform a second cerebral angiogram as the presence of previously not identified bdAVF was felt to be the most likely diagnosis; however, it also failed to demonstrate it.Reference Geibprasert, Pongpech, Jiarakongmun, Shroff, Armstrong and Krings1 Detailed review of both angiographies was undertaken and revealed unusual tortuous cortical veins in the left frontal lobe consistent with “pseudophlebitic” pattern seen in bdAVFs.Reference Geibprasert, Pongpech, Jiarakongmun, Shroff, Armstrong and Krings1 It was previously reported that in small number of patients bdAVFs can spontaneously close off during an acute venous hypertensive event and thus cannot be demonstrated on follow-up cerebral angiograms.Reference Kim, terBrugge, Krings, Willinsky and Wallace3 We believe that this is what happened in our patient, and spontaneous closure of his bdAVF explained the resolution of the left frontal mass lesion on follow-up MRI after brain edema has resorbed. It was also noticed that there was definite decreased perfusion of the left cavernous sinus on both angiographies (Figure 1G). When all imaging was reexamined, subtle left cavernous sinus thrombosis (CST) was seen in all previous studies (Figure 1H). Full hypercoagulability workup revealed significantly elevated titers of anticardiolipin IgG antibodies (46 CU, normal <20), which were even higher on repeat testing performed 1 month later. Hypercoagulable state due to anticardiolipin antibody syndrome was felt to be the most likely cause of long-standing left CST. Treatment with oral steroids was discontinued; and at the last follow-up 4 years after presentation, visual acuity was hand motions in the affected eye with the atrophic-looking optic nerve. There were no new brain lesions on follow-up neuroimaging.
BdAVFs account for 10%–15% of all intracranial arteriovenous malformations. They represent an abnormal communication between an artery and small veins in the dura mater.Reference Geibprasert, Pongpech, Jiarakongmun, Shroff, Armstrong and Krings1 Venous drainage of bdAVF dictates their clinical presentation: frontal bdAVFs draining toward the cavernous sinus exhibit features of orbital congestionReference Pandey, Steinberg, Westbroek, Dodd, Do and Marks4 but bdAVFs draining anteriorly have aggressive natural history with annual event rate of 15%.Reference Geibprasert, Pongpech, Jiarakongmun, Shroff, Armstrong and Krings1 Absence of dural sinus in the anterior cranial fossa causes venous drainage to flow directly into the intracranial cortical veins with retrograde venous reflux into brain parenchyma,Reference Pandey, Steinberg, Westbroek, Dodd, Do and Marks4 seen radiologically as “pseudophlebitic” pattern.
The most sensitive test for localizing arteriovenous shunting is cerebral angiography. However, its false negative rate is 10%; and in 7% of cases, fistulas can spontaneously thrombose during an acute venous hypertensive event thus no arteriovenous shunting can be demonstrated.Reference Geibprasert, Pongpech, Jiarakongmun, Shroff, Armstrong and Krings1, Reference Kim, terBrugge, Krings, Willinsky and Wallace3
We hypothesize that in our case there was a long-standing left frontal lobe bdAVF based on the presence of indirect neuroimaging findings: enlarged anterior falcine artery (branch of anterior ethmoid artery which is the most commonly reported feeder vessel in frontal bdAVFs) on cerebral angiogram plus a unique pattern of venous drainage seen in the left frontal lobe.Reference Inoue, Tagawa and Kumon5 Both brain MRI and cerebral angiogram demonstrated “pseudophlebitic” pattern implying the presence of retrograde leptomeningeal venous reflux. We believe that hyperintense white matter lesions limited to the left frontal lobe were a consequence of microischemic injury which has been reported in bdAVF.Reference Willinsky, Terbrugge, Montanera, Mikulis and Wallace6 While hypercoagulable state due to elevated plasma titers of anticardiolipin’s antibodies was likely the cause of CVT in our case (the presence of anticardiolipin antibodies has been well described to be associated with CVT), it is unclear whether bdAVF predisposes to CVT or whether bdAVF results from CVT.Reference Carhuapoma, Mitsias and Levine7, Reference Boggild, Sedhev, Fraser and Heron8
We hypothesize that the left frontal lobe bdAVF drained both posteriorly toward the cavernous sinus and anteriorly toward the frontal lobe. A hypercoagulable state conferred by the presence of very elevated anticardiolipin antibody titers likely led to the formation of left CST. This in turn resulted in an optic neuropathy by diverting venous drainage from the left optic nerve sheath that was destined to flow back into the cavernous sinus to instead flow toward the left frontal lobe, which was coincidentally chronically under high venous pressure because of the presence of bdAVF. This led to the continuous optic nerve leakage and caused massive optic nerve head edema. We hypothesize that no other signs of orbital congestion were seen because of the compartmentalization of cavernous sinus that resulted in subtle CST that affected only venous drainage from the left optic nerve sheath.
Two and a half years after the onset of left optic neuropathy, the patient had a presumed acute venous hypertensive event (clinically manifesting as a seizure) in the area supplied by bdAVF. Coincidentally, elevated titers of anticardiolipin antibodies conferred a prothrombotic state causing spontaneous clotting of the bdAVF during the acute event; thus, no shunting of bdAVF was demonstrated on two cerebral angiograms. Closure of bdAVF then led to the resorption of brain edema and dramatic resolution of frontal lobe lesion.
This unique case demonstrates how interaction between hypercoagulable state and the presence of bdAVF in the same patient led to monocular visual loss but serendipitously caused spontaneous resolution of bdAVF.
Disclosures
The authors have no conflicts of interest to declare.
Statement of Authorship
All authors contributed equally to the preparation of the manuscript.

