


Introduction
Pseudobulbar affect (PBA) is a disorder of emotional expression that occurs secondary to neurologic disorders or injuries affecting the brain and is characterized by frequent, disruptive, and uncontrollable outbursts of laughing and/or crying that are independent of mood and out of proportion or incongruous with social context.Reference Schiffer and Pope 1 – Reference Wortzel, Oster, Anderson and Arciniegas 3 PBA is believed to arise from midline or bilateral brain injury, degenerative processes, or lesions involving pathways that regulate affect or emotional expression. Lesions of various etiologies (inflammatory, ischemic, hemorrhagic, or neurodegenerative) involving the corticobulbar tracts, basal ganglia, and multiple, bilateral cortical structures have been associated with the uncontrollable laughing or crying of PBA.Reference Parvizi, Coburn, Shillcutt, Coffey, Lauterbach and Mendez 4 , Reference Brooks, Crumpacker, Fellus, Kantor and Kaye 5 PBA episodes are usually upsetting and embarrassing for patients and family members and may adversely affect life situations.Reference Schiffer and Pope 1 – Reference Wortzel, Oster, Anderson and Arciniegas 3 , Reference Colamonico, Formella and Bradley 6 Patients have reported that laughing and crying episodes have contributed to job loss, isolation from friends and family, and even nursing home placement.Reference Colamonico, Formella and Bradley 6 Published estimates of PBA symptom prevalence in dementia range from 9% to 39% depending on definition and method of ascertainment.Reference Brooks, Crumpacker, Fellus, Kantor and Kaye 5 , Reference Work, Colamonico, Bradley and Kaye 7 , Reference Starkstein, Migliorelli and Teson 8 However, the condition may go undiagnosed, possibly due to low awareness by patients and caregivers and diagnostic confusion with depression or other neuropsychiatric conditions.Reference Wortzel, Oster, Anderson and Arciniegas 3 , Reference Work, Colamonico, Bradley and Kaye 7
PBA diagnosis is based on clinical history and neurologic examination. Key features include uncontrollable, stereotypical laughing and/or crying episodes that can last seconds to several minutes, occur multiple times per day, and result in clinically significant distress.Reference Cummings, Arciniegas and Brooks 2 PBA episodes are typically triggered by mildly emotional events that would not ordinarily elicit a profound response. Signs of pseudobulbar palsy (hyperactive jaw jerk, exaggerated gag reflex, tongue weakness, dysarthria, and dysphagia)Reference Cummings, Arciniegas and Brooks 2 may be present but are not required for a diagnosis.Reference Cummings, Arciniegas and Brooks 2 , Reference Parvizi, Arciniegas and Bernardini 9 The principal differential diagnosis of PBA with crying episodes is depression. However, unlike depression, patients with PBA are not necessarily sad, nor do they cry because of depressed mood. Conversely, clinical depression is not characterized by frequent stereotypical crying episodes. In patients with dementia, where laughing or crying episodes and associated mood state may be difficult to discern because of cognitive impairment, establishing a PBA diagnosis may be reliant on caregiver reports, the observable characteristics of crying and/or laughing spells, the relationship of episodes to social context, and the level of disruption caused by the episodes.
The fixed combination of dextromethorphan and quinidine (DM/Q; NUEDEXTA® [Avanir Pharmaceuticals, Inc., Aliso Viejo, CA, USA])Reference Pioro, Brooks and Cummings 10 , 11 is the only therapy with regulatory approval for PBA treatment. Although antidepressants have shown effectiveness in studies evaluating their use for treatment of PBA-like symptoms, the level of clinical evidence supporting their use is suggestive, not definitive, and mostly has been confined to studies of patients with emotionalism, with or without depression, following stroke. A Cochrane Review concluded that although some studies of antidepressants showed large treatment effects, confidence intervals were wide and methodological difficulties prevented definitive recommendations regarding their use.Reference Hackett, Yang, Anderson, Horrocks and House 12 Methodological difficulties of these studies have included the use of small sample sizes, lack of adequate treatment concealment, lack of standardization of measures to diagnose PBA or its change over time with treatment, depression as a confounding condition, and lack of systematic collection of adverse event data.Reference Hackett, Yang, Anderson, Horrocks and House 12 – Reference Pioro 14 Dextromethorphan, the active component of DM/Q in the central nervous system, is pharmacologically a weak, uncompetitive N-methyl-d-aspartate receptor antagonist, a moderate-affinity sigma-1 receptor agonist, a serotonin and norepinephrine reuptake inhibitor, and an α3β4 neuronal nicotinic receptor antagonist.Reference Hernandez, Bertolino, Xiao, Pringle, Caruso and Kellar 15 – Reference Werling, Keller, Frank and Nuwayhid 17 Quinidine is a potent inhibitor of cytochrome P450 2D6 (CYP2D6). Dextromethorphan typically has poor central nervous system availability owing to rapid CYP2D6 metabolism to dextrorphan; however, in the presence of low-dose Q (10 mg), systemic exposure to DM is elevated approximately 20-fold and dextrorphan exposure is reduced, 11 thereby altering the pharmacokinetic and pharmacodynamic profile of DM and enabling therapeutic central nervous system exposure. In a controlled clinical trial, DM/Q was significantly more effective at treating PBA symptoms than DM or Q administered alone.Reference Brooks, Thisted and Appel 18
Controlled studies leading to DM/Q approval were conducted in patients with PBA secondary to amyotrophic lateral sclerosis (ALS) or multiple sclerosis (MS).Reference Pioro, Brooks and Cummings 10 , Reference Brooks, Thisted and Appel 18 , Reference Panitch, Thisted and Smith 19 Additionally, a large (N=553), 52-week safety trial of a higher DM/Q dose enrolled patients with PBA across multiple etiologies but only a relatively small number of patients with dementia (n=17), stroke (n=51), or traumatic brain injury (TBI; n=23).Reference Pattee, Wymer, Lomen-Hoerth, Appel, Formella and Pope 20 Since DM/Q became commercially available in 2010, an estimated 42,000 patient-years of exposure across distinct patient populations with PBA have provided information on DM/Q safety; nonetheless, data from more controlled clinical experience in specific populations are desirable.
The Pseudobulbar Affect Registry Investigating Symptom Management II (PRISM II) trial was conducted to gain additional safety, tolerability, and effectiveness data on DM/Q for the treatment of patients with PBA secondary to dementia, stroke, or TBI, thus expanding the evidence with DM/Q in 3 additional neurologic conditions commonly associated with PBA. The PRISM II dementia cohort was completed on July, 15, 2014, and the results are reported here. The stroke and the TBI cohorts are ongoing and will be reported separately.
Methods
Study design
PRISM II (clinicaltrials.gov identifier: NCT01799941) was an open-label, 90 Day, US-based multicenter trial enrolling adults aged ≥18 years with PBA secondary to dementia, stroke, or TBI. The study was conducted according to Good Clinical Practice and the Declaration of Helsinki. All patients received DM/Q 20/10 mg twice daily (once daily during Week 1). Clinic visits occurred at baseline and at Days 30 and 90 or early termination, with a phone visit at Day 60.
Patients
Patients were eligible for enrollment if they had a clinical diagnosis of dementia (including Alzheimer’s disease [AD] or vascular, Lewy body, or frontotemporal dementia), and a clinical diagnosis of PBA based on published criteria.Reference Cummings, Arciniegas and Brooks 2 PBA was defined by episodes of involuntary or exaggerated emotional expression (ie, laughing or crying) that resulted from a brain disorder, represented a change from the person’s usual emotional reactivity, were incongruent with or in excess of the person’s corresponding mood state, and were independent or in excess of any provoking stimulus, with symptoms not being better accounted for by another neurologic or psychiatric disorder or the direct physiologic effect of substance abuse or medication. Patients were also required to have a Center for Neurologic Study–Lability Scale (CNS-LS) score ≥13Reference Moore, Gresham, Bromberg, Kasarskis and Smith 21 and a stable, non-rapidly changing neurologic condition for ≥3 months preceding enrollment. Stable doses (≥6 weeks) of memantine or acetylcholinesterase inhibitors and stable doses (≥2 months) of antidepressants or medications for affective/behavioral or emotional symptoms were allowed. All patients (or authorized individuals) provided written informed consent. Patients who were unable to complete study measures were required to have a caregiver who could complete the measures on their behalf; a caregiver was defined as a person who spent ≥3 days of waking hours with the patient for the week prior to clinic visits (to be knowledgeable about PBA episodes).
Patients were excluded if they had severe dementia (Mini-Mental State Examination [MMSE] score <10), a recent stroke (within 3 months), penetrating TBI, severe depressive disorder, psychosis or bipolar disorder (or were residing in a mental health facility), unstable systemic disease, a very short life expectancy (≤6 months), or any labeled contraindication to DM/Q use (including QT interval prolongation). The use of DM/Q in the past 6 months, substance/alcohol abuse in the past 3 years, or participation in an interventional clinical study within the past 30 days were also exclusionary.
Outcome measures
Primary measure
The primary effectiveness outcome was change in CNS-LSReference Moore, Gresham, Bromberg, Kasarskis and Smith 21 score from baseline to Day 90/final visit. The CNS-LS is a 7-item, self-rated questionnaire that has been validated in patients with ALSReference Moore, Gresham, Bromberg, Kasarskis and Smith 21 and in patients with MSReference Smith, Berg, Pope, Callahan, Wynn and Thisted 22 as a quantitative measure of frequency and severity of PBA laughing and crying episodes. Four questions measure labile laughter and 3 measure labile tearfulness, with scores ranging from 7 (no symptoms) to 35 (maximum symptom severity and frequency). This scale is sensitive to change over time and sensitive to the effects of treatment, and score change corresponds well with change in PBA episodes.Reference Brooks, Thisted and Appel 18 , Reference Panitch, Thisted and Smith 19 , Reference Moore, Gresham, Bromberg, Kasarskis and Smith 21 In this study, the CNS-LS was completed by the patient (or caregiver as a patient proxy) at baseline, Day 30 (visit 1), and Day 90 or early withdrawal (final visit).
Secondary measures
Secondary effectiveness measures included change from baseline in number of PBA laughing or crying episodes per week (estimated for the 7 days before each clinic visit); Clinical Global Impression of Change (CGI-C) and Patient Global Impression of Change (PGI-C), rated at Day 90/final visit by the investigator and patient (or caregiver), respectively, assessing overall change in the patient’s condition (with respect to PBA) on a 7-response Likert-type scale (1=very much improved to 7=very much worse); change from baseline to Day 90/final visit on a quality-of-life visual analog scale (QOL-VAS), consisting of an anchored, continuous line scale that assessed the impact of PBA episodes on the patient’s global subjective well-being (0=not at all affected to 10=significantly affected) during the past week; and a single question measuring patient satisfaction with treatment, rated at Day 90/final visit on a 5-point scale (1=very dissatisfied to 5=very satisfied). Investigators also administered the MMSE at baseline and Day 90/final visit,Reference Folstein, Folstein and McHugh 23 which is composed of 11 questions or simple tasks assessing orientation, memory, attention, and language to evaluate the patient’s cognition (scored from 0 to 30). Additionally, patients or caregivers completed the Patient Health Questionnaire (PHQ-9), a 9-item scale assessing depressive symptoms at baseline, Day 30, and Day 90/final visit. The PHQ-9 is scored from 0 to 27, with higher scores indicating more severe depression.
Safety
Safety was assessed using adverse events (AEs) recorded throughout the study and clinical assessments during study visits.
Statistical analysis
The population defined for effectiveness analyses consisted of all patients who met inclusion criteria, received at least 1 dose of DM/Q, and had at least 1 post-baseline CNS-LS score. The safety population comprised all patients who received at least 1 dose of DM/Q. All data were analyzed descriptively. Effectiveness ratings at Day 30 and/or Day 90/final visit compared with baseline were also analyzed inferentially using 1-sample t-tests for rating scale measures (CNS-LS, QOL, MMSE, and PHQ-9) and a mixed-effects Poisson regression model to estimate change in PBA episode counts. The mixed-effects Poisson regression model incorporated age, gender, and time (Day 30 and Day 90) as fixed effects while allowing for individual differences in baseline episode count (a random subject effect). The percentage change in episode rate from baseline to any visit is 1 minus the appropriate time parameter (λ).
The primary analysis tested the null hypothesis that the mean change in CNS-LS score from baseline to the Day 90/final visit was equal to 0; the 95% confidence interval (CI) was also reported to enable a descriptive comparison with the CNS-LS change in the phase 3, pivotal registration trial that led to US approval of DM/Q for PBA.Reference Pioro, Brooks and Cummings 10 Additional analyses included percentage of patients experiencing PBA episode remission; correlation analysis between CNS-LS score and PBA episode count and between these measures and other validated instruments at each time point; and analysis of outcomes based on respondent type (those obtained from patients vs. from caregiver proxies). Tests of significance were 2-tailed and carried out at the α=0.05 level of significance; all analyses were completed using SAS v9.2 (SAS Institute Inc., Cary, NC, USA) or Stata v12 (StataCorp, College Station, TX, USA). All patients with complete data for the given comparison were included.
A power calculation was conducted using results from the pivotal DM/Q phase 3 trial,Reference Pioro, Brooks and Cummings 10 in which mean (SD) change from baseline to Day 84 in CNS-LS was –8.2 (6.1) points for DM/Q and –5.7 (5.3) points for placebo. Assuming a SD of 6.1, and based on the use of a 2-sided, 1-sample t-test at the α=0.05 level of significance, a sample size of 100 subjects in each of the 3 clinical diagnosis cohorts would provide 80% power to detect improvement of 1.75 points from an assumed true placebo mean change of –5.7 points (ie, a mean change of –7.45 points) and 90% power to detect an improvement of 2.0 points over the assumed true placebo mean change (ie, a mean change of –7.7 points). The assumptions of magnitude of effect and SD for the power calculation were supported by an interim analysis of this study following completion of the first 100 subjects (regardless of cohort), and determined that approximately 100 patients in each disease cohort would provide sufficient power to meet the protocol-specified endpoints and, therefore, the dementia cohort was subsequently closed with 134 patients enrolled.
AEs were categorized via Medical Dictionary for Regulatory Activities (MedDRA version 15.1) coding and reported descriptively.
Results
Patient disposition and baseline characteristics
Of the 143 patients with dementia screened, 134 were enrolled in PRISM II, received at least 1 dose of DM/Q, and were included in the safety population. Of these 134 patients, 106 (79.1%) completed the study and 28 (20.9%) terminated participation before completion (Figure 1). The most common reasons for early termination were AEs (10.4%) and withdrawal of consent (5.2%). A total of 26 (19.4%) enrolled patients were excluded from effectiveness analyses, 16 because they did not have a post-baseline CNS-LS score and 10 because they did not meet all study eligibility criteria, leaving 108 (80.6%) patients to compose the effectiveness population. Baseline demographic and clinical characteristics for the enrolled (safety) population (n=134) are summarized in Table 1. The mean (SD) patient age was 70.7 (12.1) years, with 43.3% of patients aged ≥75 years; 59.0% were female, 88.1% were white, 25.4% were Hispanic/Latino, 35.1% lived in a skilled nursing or assisted living facility, and 73.1% had a caregiver. The most common dementia diagnosis was probable AD (64.2%), followed by vascular (15.7%), frontotemporal (9.0%), and Lewy body dementia (3.7%); 7.46% had “other” dementia diagnoses. Most patients (93.3%) were assessed as having dementia of mild to moderate severity. Overall, patients were taking a median of 8 (range 0–27) medications at study entry, with 81.3% taking at least 1 psychopharmacologic medication, most commonly antidepressants (56.7%), benzodiazepines (34.3%), and antipsychotics (29.1%). Medications for dementia were used by 54.5%, including acetylcholinesterase inhibitors (46.3%) and memantine (28.4%).
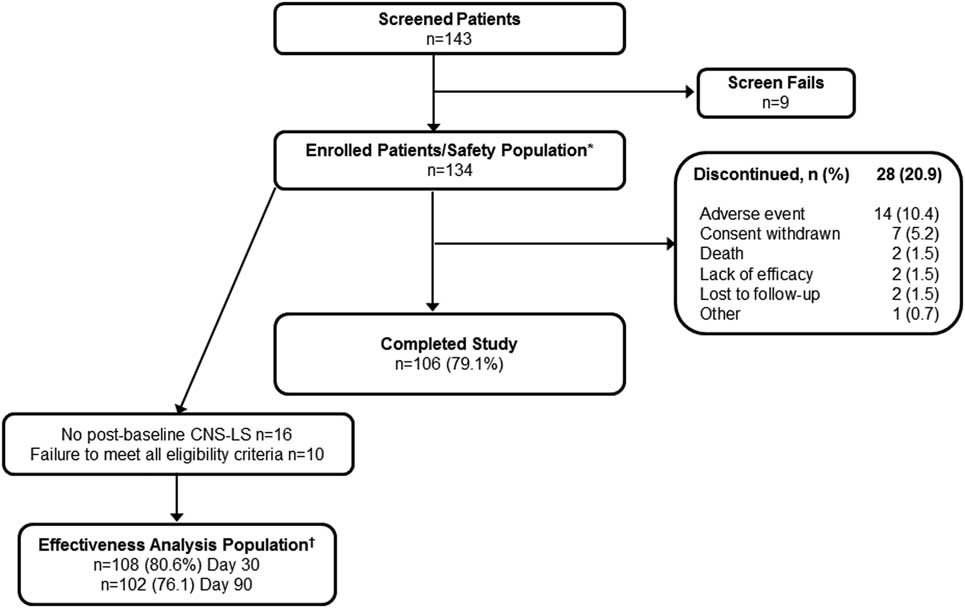
Figure 1 Patient disposition: dementia cohort of PRISM II. *Safety population consisted of all enrolled patients who received at least 1 dose of DM/Q. †Effectiveness analysis population consisted of patients meeting all inclusion criteria, including a CNS-LS score of ≥13, who received at least 1 dose of DM/Q and had a CNS-LS score at Day 30 or Day 90/Final Visit. CNS-LS, Center for Neurologic Study–Lability Scale; DM, dextromethorphan; Q, quinidine.
Table 1 Baseline demographics and clinical characteristics

a Other dementia included dementia due to multiple sclerosis (n=4), Parkinson’s disease (n=1), alcohol-related and vascular (n=1), “brain cell deterioration” (n=1), “subcortical alterations” (n=1), unspecified causes (n=1), and mild cognitive impairment (n=1).
b As rated by investigators.
c Psychopharmacologic medications included antipsychotics, antidepressants, sedative/hypnotics, anxiolytics and other benzodiazepines such as clonazepam.
d Other antidepressants include bupropion, duloxetine, trazodone, mirtazapine, venlafaxine, milnacipran, and vilazodone
e The CNS-LS scale ranges from 7 (no symptoms) to 35 (maximum symptom frequency and severity).
f Effectiveness analysis population (n=108).
CNS-LS, Center for Neurologic Study-Lability Scale; MMSE, Mini-Mental State Examination; PBA, pseudobulbar affect; SD, standard deviation; SSRIs, selective serotonin reuptake inhibitors.
Effectiveness measures
Primary effectiveness endpoint
The CNS-LS score showed progressive and statistically significant improvement compared with the baseline mean (SD) score of 20.1 (4.2), with reductions at Day 30 of –4.6 (5.1) and at Day 90/Endpoint of –7.2 (6.0) [P<.001 for both; Figure 2]. This CNS-LS reduction at Day 90/Endpoint fell within the 95% CI for CNS-LS reduction seen with DM/Q (95% CI –9.48, –7.00) in the phase 3 pivotal trial conducted in patients with PBA secondary to ALS or MS and was below the lower limit of the 95% CI for the CNS-LS reduction seen with placebo in that trial (95% CI –6.77, –4.68).
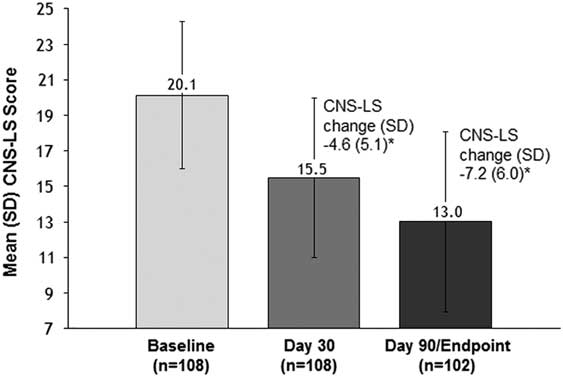
Figure 2 Mean (SD) CNS-LS score at baseline, Day 30, and Day 90 (effectiveness analysis population, dementia cohort). CNS-LS scores range from 7 (normal) to 35 (maximum frequency and severity). P values are based on the one sample t-test and represent comparison to baseline. *P<.001 vs. baseline. CNS-LS, Center for Neurologic Study–Lability Scale; PBA, pseudobulbar affect; SD, standard deviation.
Secondary effectiveness analyses
The median (min, max) PBA episodes/week decreased from 21 (0, 90) at baseline to 6 (0, 77) at Day 30 and 3 (0, 80) at Day 90/Endpoint. The frequency distributions of PBA episodes at baseline, Day 30, and Day 90/Endpoint, as well as the model-based estimates, are shown in Figure 3. Overall, the estimated number of PBA episodes in the week before assessment was reduced by 50.2% at Day 30 and 67.7% at Day 90/Endpoint compared with baseline (P<.001 for both; mixed-effects Poisson regression model). Remission of PBA symptoms (defined as no reported episodes in the week before assessment) was observed in 13.0% of patients at Day 30, and 31.4% of patients at Day 90/Endpoint. On CGI-C and PGI-C, 77.5% (n=79/102) and 76.5% of patients (n=78/102), respectively, were rated as much improved or very much improved at Day 90/Endpoint (Figure 4). Mean (SD) QOL-VAS scores improved from 6.0 (2.78) at baseline to 2.7 (2.36) at Day 90/Endpoint (P<.001). On the Patient Treatment Satisfaction Survey, 74.5% of patients or their caregivers were somewhat satisfied (21.6%) or very satisfied (52.9%) with treatment, while 11.8% were somewhat dissatisfied (6.9%) or very dissatisfied (4.9%), with the remaining 13.7% reporting a neutral response. Mean (SD) MMSE scores improved by 0.54 (3.14) points, from 20.2 (5.57) at baseline to 21.0 (6.36) at Day 90/Endpoint (P=.084). Mean (SD) PHQ-9 scores improved from 13.2 (5.26) at baseline to 7.4 (5.19) at Day 90/Endpoint (P<.001).
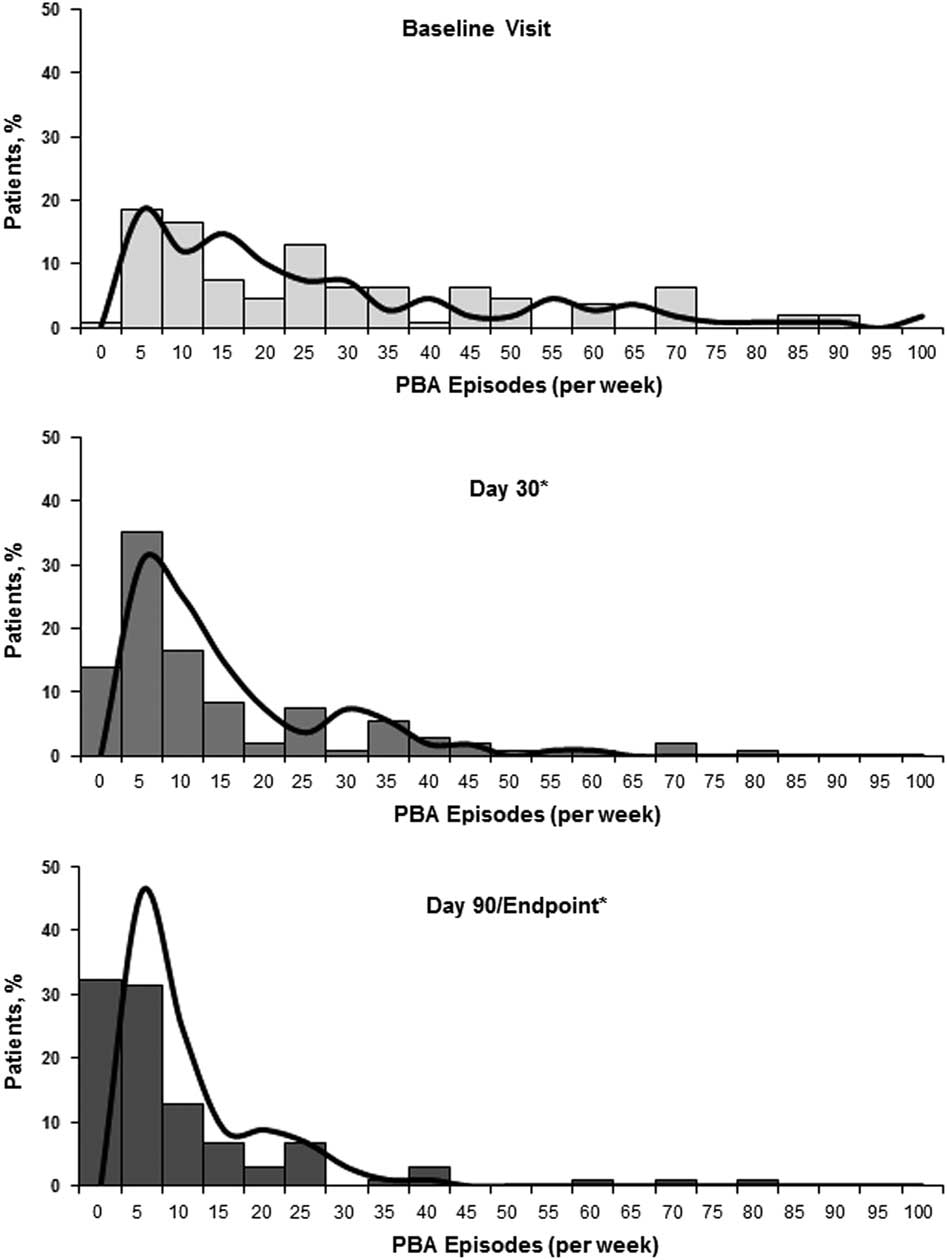
Figure 3 Distribution of PBA episodes (per week) by visit. Solid bars illustrate the percentage of patients experiencing the given number of episodes shown within the range provided; the x-axis data label represents the maximum of the range for episodes/week in blocks of 5 (eg, at baseline 18.5% of patients had 1-5 PBA episodes/week; 16.7% had 6-10 episodes/week, etc.). The solid curved line represents the number of PBA episodes that would be predicted based on each patient’s values for the parameters (age, gender, and time [Day 30, Day 90]; fixed effects) and baseline rate (random-effects) in the mixed effects Poisson regression model. Patients or daytime caregivers were asked to estimate the total episodes of exaggerated or uncontrollable laughing and/or crying over the past week (prior to visit) at baseline, 30 days, and 90 days. Estimated percent change from baseline for PBA episode count was evaluated via a mixed effects Poisson regression model for the effectiveness analysis population. *P<.001 vs. baseline. PBA, pseudobulbar affect.
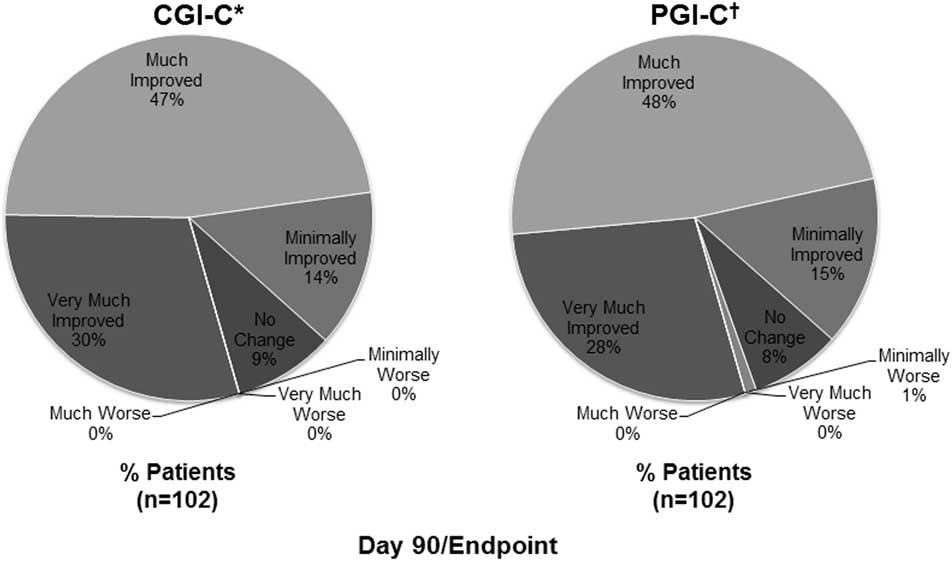
Figure 4 Clinical and patient global impression of change (effectiveness analysis population). *CGI-C is a 7-point investigator-rated scale that assessed the patient's overall treatment response (with respect to PBA) from baseline to Day 90/Endpoint, rated as very much improved, much improved, minimally improved, no change, minimally worse, much worse, or very much worse. †PGI-C is a 7-point patient [or the patient's caregiver]–rated scale that assessed overall treatment response (with respect to PBA) from baseline to Day 90/Endpoint, rated as very much improved, much improved, minimally improved, no change, minimally worse, much worse, or very much worse. CGI-C, Clinical Global Impression of Change; PBA, pseudobulbar affect; PGI-C, Patient Global Impression of Change.
Relationship of patient and caregiver ratings
Approximately 40% of ratings were completed by caregivers, and, in most cases (~90%), the rater (patient or caregiver) did not change from baseline to endpoint. Compared with patient-respondents, caregiver-respondents generally reported a greater PBA symptom change from baseline to endpoint (mean [SD] CNS-LS reduction of –6.20 [6.16; n=56] vs. –8.16 [4.94; n=37] from a baseline of 19.8 [3.69; n=57] vs. 20.1 [4.70; n=41] and PBA episode reduction of 57.7% vs. 77.2% from a baseline of 23.8 [21.83; n=53] vs. 27.7 [23.21; n=43]). Patient vs. caregiver responses were statistically significantly different from each other only for estimated PBA episode count reduction (P<.001) but not for changes in CNS-LS or other study effectiveness outcomes.
Safety
A total of 49 (36.6%) of the 134 patients who received DM/Q reported at least 1 treatment-emergent AE; the most commonly reported AEs were headache (7.5%), urinary tract infection (4.5%), and diarrhea (3.7%) (Table 2). Most treatment-emergent and treatment-related AEs were mild or moderate in intensity. Fourteen (10.4%) patients experienced a serious AE, including 2 patients who died during the study: a 91-year-old man with a history of coronary artery disease, atrial fibrillation, and hypertension died on Study Day 10 of a myocardial ischemic event, and an 83-year-old man died of suspected cerebrovascular accident and cardiac arrest on Study Day 31. Both were deemed not related to study drug after careful review by the investigators. None of the other serious AEs were considered related to DM/Q treatment, and no serious AE type (by preferred term) was reported in more than 1 patient each. Sixteen (11.9%) patients had AEs that led to discontinuation, including the 2 patients who died.
Table 2 Summary of adverse events (AEs) in the dementia cohort of PRISM II

AE, adverse event.
Discussion
The PRISM II open-label study assessed the effectiveness of DM/Q for PBA in patients with dementia, stroke, and TBI. The results for the dementia cohort reported here represent the first prospectively conducted, systematic study of PBA treatment in patients with dementia, and they add to the evidence supporting DM/Q effectiveness for PBA across different neurologic conditions where PBA may occur. The significant improvement in PBA symptoms, as shown by the reductions in CNS-LS score, measuring episode frequency and severity, and PBA episode rate were clinically meaningful as reflected by clinician and patient/caregiver ratings of global improvement and improvement in a QOL measure based on the specific impact of PBA. These improvements were noted in a patient population where over half were already receiving antidepressants and 29% were receiving antipsychotics. DM/Q was also well tolerated in this elderly population, most of whom were receiving several concomitant medications (median 8 per patient), including 46% taking acetylcholinesterase inhibitors and 28% receiving memantine. Serious AEs were not thought to be related to study treatment, and few patients discontinued the study as a result of AEs. Treatment-emergent AEs were consistent with the known safety profile of DM/Q. 11 The most frequently reported AEs in this trial—diarrhea, dizziness, and urinary tract infection—occurred at a greater incidence with DM/Q than with placebo in the pivotal trial in patients with PBA secondary to ALS or MS; however, headache, nausea, fall, and somnolence occurred at a lower incidence with DM/Q than with placebo in that trial.Reference Pioro, Brooks and Cummings 10 Mean MMSE scores increased slightly over the course of the trial, indicating a lack of adverse effect on cognitive function.
Limitations of this study include those related to its open-label design, which allows for the possibility that observations may be influenced by bias on the part of the patient, caregiver, or investigator. However, results from this trial were consistent with those of the pivotal 12-week, placebo-controlled trial in patients with PBA secondary to ALS and MS.Reference Pioro, Brooks and Cummings 10 Another potential limitation is the fact that the CNS-LS was not previously validated as a measure of PBA symptoms in patients with dementia. However, baseline CNS-LS scores were consistent with baseline scores reported in other PBA clinical trials using a similar clinical diagnosis of PBA for study inclusion, and improvement in CNS-LS scores correlated with improvements in other measures performed by the investigator and caregiver, including PBA episode count, CGI-C/PGI-C with respect to PBA symptoms, and QOL-VAS ratings. Similar correlations were also observed in the previous controlled trials of DM/Q for PBA.Reference Brooks, Thisted and Appel 18 , Reference Panitch, Thisted and Smith 19 Also, although it is not uncommon for a study to assess adverse events, vital signs, and other investigator-reported measures without a structured safety assessment scale, this can, perhaps, be perceived as a limitation. Further, the lack of a placebo arm limits the ability to compare reported adverse events against a control group. Still, safety findings were consistent with previously published DM/Q studies. Finally, patients with dementia may be poor informants for certain measures (ie, PBA episode recall). A comparison of ratings showed numerically larger changes for all measures except PHQ-9 (comparable) when the caregiver completed the rating on the patient’s behalf; however, these differences reached significance only for PBA episode count (reduction from baseline of 77.2% based on caregiver-respondent reports vs. 57.7% for patient-respondent reports; P<.001). This analysis would suggest that the overall magnitude of change in effectiveness outcomes was not exaggerated due to poor patient recall.
Conclusion
This study demonstrated that twice daily DM/Q 20/10 mg reduced the frequency and severity of PBA symptoms in a cohort of patients with dementia as measured by the CNS-LS, the number of PBA episodes, and global measures of clinical change. This trial provides prospectively collected information on the use of DM/Q for the treatment of PBA using standard, validated clinical measurements in patient populations where limited clinical trial data were available, including elderly (mean age in this cohort was 71 years) and cognitively impaired persons. The AE profile observed with DM/Q was consistent with that previously reported for this drug combination and with the health status of this elderly population with dementia. The effectiveness and safety attributes of DM/Q in this study appear comparable to phase 3 trial results in PBA secondary to ALS or MS and are supportive of DM/Q use in PBA secondary to dementing conditions. Evidence regarding DM/Q for the treatment of PBA in patients with stroke or TBI will be reported in the near future.
Disclosures
Rachelle Doody has consulted within the past 12 months for AbbVie, AC Immune, Avanir Pharmaceuticals, Inc., AZ Therapies, Baxter, Biote, CereSpir, Chiesi, GlaxoSmithKline, Hoffmann-La Roche, NeuroCog, Novartis, and Pfizer; has stock options in AZ Therapies, QR Pharma, Sonexa, and Transition; receives funding from the NIH Alzheimer’s Disease Cooperative Study and from the Texas Alzheimer’s Research and Care Consortium; serves as principal investigator on clinical trials funded by Accera, Avanir, Genentech, Janssen Alzheimer Immunotherapy, NIH, Pfizer, and Takeda; and serves on the editorial boards of Alzheimer’s Research and Therapy and Dementia and Geriatric Cognitive Disorders. Stephen D’Amico has received honoraria as a consultant and speaker for Avanir Pharmaceuticals, Inc. and has been a consultant and received research grants from Sanofi, Merck, AstraZeneca, Bristol-Myers, Novartis, and Takeda Pharmaceuticals. Andrew Cutler has served as a consultant for, received research grants from, and served as a speaker for Abbott, AstraZeneca, Bristol-Myers Squibb, Forest, GlaxoSmithKline, Lilly, Merck, Novartis, Ortho-McNeil-Janssen, Otsuka, Pamlab, Pfizer, Shire, Sunovion, Takeda, and Vanda. Charles Davis is a consultant to Avanir Pharmaceuticals, Inc. Paul Shin, Fred Ledon, Charles Yonan, and João Siffert are employees of Avanir Pharmaceuticals, Inc.
Acknowledgments
The authors acknowledge William Sauve, MD, Flora Hammond, MD, Richard Zorowitz, MD, and David Alexander, MD (protocol development); Shereen McIntyre, MBA (clinical data management and data analysis); Tracy Maines and Yim Ang (project management); Andrea Formella, PharmD, BCPP (protocol development, data analysis, and editorial assistance); Jennifer Lee (research coordinator); Randall Kaye, MD (study concept design); Shelby Woods (publication management); Rachel Halpern, PhD, and Mike Johnson, MS (statistical analysis) for their contributions. Medical writing assistance was provided by John H. Simmons, MD, of Peloton Advantage, LLC, and was supported by Avanir Pharmaceuticals, Inc. The authors are entirely responsible for the scientific content of this manuscript.
This study was funded by Avanir Pharmaceuticals, Inc. Rachelle Doody wrote the first draft of the manuscript. John H. Simmons, MD, of Peloton Advantage, LLC, was supported by Avanir Pharmaceuticals, Inc., to prepare the revised drafts of this manuscript according to the suggestions from authors. All authors had full access to the study data and had final responsibility for the decision to submit for publication. All authors approved the final version.







 Open access
Open access