

INTRODUCTION
Staphylococcus aureus is a leading cause of nosocomial and community infections around the world [Reference Lowy1] and is associated with several different infections [Reference Diekema2]. The emergence and spread of multiresistant strains, and particularly of methicillin-resistant S. aureus (MRSA) has made the treatment of staphylococcal infections a global challenge [Reference Fuda, Fisher and Mobashery3, Reference Grundmann4].
In Italy, after an initial and continuous increase [Reference Fuda, Fisher and Mobashery3, Reference Stefani and Varaldo5, Reference D'Ancona6], the frequency of isolation of MRSA, from 2002 onwards, has undergone a constant, although small average increment, reaching values around 33% in 2007 and 2008 [European Antimicrobial Resistance Surveillance (EARSS) annual report 2008 at http://www.earss.rivm.nl]. Most MRSA are isolated from subjects aged >65 years, who are frequently hospitalized, in intensive care and/or following surgery [Reference Fuda, Fisher and Mobashery3, Reference D'Ancona6]. A recent study [Reference Campanile7] suggests that, although indicative of general trends, data from the EARSS programme are only partially representative of the prevalence of nosocomial MRSA in Italy. In fact, MRSA is responsible for various infections and considerable variations exist between institutions and wards, even in the same geographical areas, probably due to local public health strategies and other variables.
Complexity in the epidemiology of infections caused by S. aureus is also the result of significant genetic diversity and clonal evolution, which was undoubtedly accelerated by the strong selective pressure due to the widespread use of antibiotics that eventually broke interlineage barriers allowing the acquisition and/or loss of virulence genes transported by mobile genetic elements [Reference Argudìn8].
Resistance to methicillin is normally dependent on the presence of the mecA gene, encoding a penicillin binding protein (PBP) showing low-affinity for β-lactams [Reference Fuda, Fisher and Mobashery3]. The mecA gene is inserted into the staphylococcal chromosomal cassette (SCCmec), which is incorporated into the chromosome of S. aureus at a specific site [Reference Katayama, Ito and Hiramatsu9, Reference Kondo10]. Currently, seven types of SCCmec (I–VII) and an increasing number of variants, have been described [Reference Higuchi11]. Besides differences in essential genetic components, the various SCCmec elements differ from each other in that they can host a variety of accessory resistance genes involved in resistance to non-β-lactam antibiotics. In fact, in contrast with SCCmec types II and III that contain many antibiotic and heavy-metal resistance genes in integrated plasmids or transposons [Reference Grundmann4] other SCCmec types, with some exceptions, do not contain any antibiotic resistance genes other than mecA. In the Italian study cited previously [Reference D'Ancona6], 54·6% of the isolates were resistant to >4 antibiotics, suggesting a large diffusion of SCCmec cassettes harbouring multi-drug resistance elements, although the presence of different determinants of resistance cannot be excluded.
MRSA strains emerged and diffused in the community (CA-MRSA) and were shown frequently to carry the smaller and more easily mobilized SCCmec cassettes and are generally less drug resistant than their counterparts acquired in hospitals (HA-MRSA) [Reference Grundmann4]. Nevertheless, CA-MRSA are becoming a serious public health concern since they generally produce a wider variety of pathogenicity and virulence factors, including the PVL toxin, and can cause different serious infections, including acute, invasive, and necrotizing syndromes, and may occasionally spread in hospitals [Reference Boyle-Vavra and Daum12]. The reasons why MRSA, after 40 years of fortuitous inability to colonize people outside the hospital setting, apparently acquired this crucial ability remain obscure [Reference Gorwitz13].
As a consequence of several important differences in pathogenicity and resistance to antimicrobials existing between HA-MRSA and CA-MRSA, the availability of simple and reliable criteria to define the nature of the infection would be of considerable importance. Currently there is some controversy in defining these two groups; distinction is generally based on temporal criteria in terms of time of MRSA isolation after hospital admission, history and time of recent hospitalization and exploitation of other risk factors, e.g. recent surgery, dialysis [Reference Maltezou and Giamarellou14]. Therefore, making an absolute distinction between HA-MRSA and CA-MRSA based on these criteria is sometimes difficult to achieve. While the frequency of CA-MRSA isolation is apparently increasing, the careful evaluation of several CA infections often reveals a history of recent hospitalization, or of close contact with hospitalized or recently hospitalized people, or the existence of other predisposing factors. Nevertheless, most of these strains show resistance patterns typical of CA-MRSA, in which β-lactam resistance predominates [Reference Maltezou and Giamarellou14] while the isolation of multidrug-resistant strains from infections arising in the community is not uncommon.
We aimed to perform surveillance of S. aureus, with particular attention to MRSA and its distribution within the nosocomial and community district in the area of Viterbo (central Italy) and the surrounding areas. The genetic diversity of MRSA isolates was analysed by simple and inexpensive molecular methods to assess if classification of MRSA as HA and CA by simple temporal criteria correlates with the genetic profiles of isolates.
METHODS
This study was conducted during the period January 2003 to June 2008 on different clinical samples submitted for routine analyses to the microbiology laboratory of Hospital ‘Belcolle’ in Viterbo, where isolation, identification and antimicrobial susceptibility testing were performed. MRSA isolates were then characterized by molecular methods at the Department of Public Health Sciences of the University of Rome ‘La Sapienza’.
Microbiological analyses
Clinical samples were obtained from patients attending the hospital itself, withdrawal centres, clinics, day hospitals, and nursing services in Viterbo and district, and processed by standard methods [Reference Kloos, Jorgensen, Lennette, Balows, Hausler and Shadomy15]. Identification of antimicrobial susceptibility of S. aureus was determined by the Vitek 2 automated system (bioMérieux, Italy). Isolates were classified as MRSA on the basis of resistance to oxacillin (⩾4 μg/ml).
An infection was considered to be hospital acquired if it developed within 48 h after hospital admission, up to 3 days after discharge from hospital, or up to 30 days following surgery.
Purification of genomic DNA
Genomic DNA was prepared from each MRSA isolate grown in LB broth overnight at 37°C. The bacterial pellet was suspended at an OD 600 nm=20 in 50 mm Tris–HCl, 145 mm NaCl (pH 7·5), containing sucrose 15% (w/v) and lysostaphin (40 U/ml) and the cell wall was digested at 37°C for 1 h. Bacterial cells were then lysed by adding 4 vol of 50 mm NaCl containing N-lauroyl-sarcosine 1% (w/v) and DNA was extracted and purified by phenol and phenol/chloroform treatments followed by precipitation with ethanol. DNA concentration was determined by the QubitTM quantitation system (Invitrogen, Italy) and samples were stored at −20°C until used.
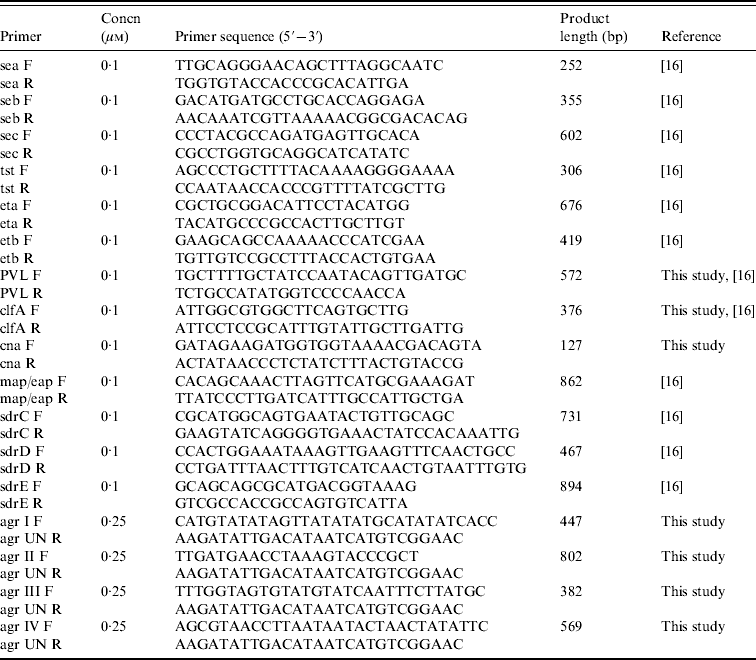
PCR assays for genotyping
Seventeen genetic determinants were assayed by PCR; these included toxins (sea, seb, sec, tst, eta, etb, PVL), adhesins (clfA, cna, map/eap, sdrC, sdrD, sdrE), and agr groups I–IV. Primers and PCR conditions were essentially as described previously [Reference Campbell16], with some modifications (Table 1). PCR amplifications were performed in a T personal thermal cycler (Biometra, Germany) with DreamTaq DNA polymerase (Fermentas, Italy). Genomic DNA (100 ng) was added to each PCR mix containing 2·5 mm MgCl2, a 10 mm concentration of the deoxynucleoside triphosphates, and the appropriate concentration of each forward and reverse primer, and Taq polymerase. Detection of genes for adhesins and toxins was performed by single PCR reactions while detection of agr group-specific sequences was performed as a multiplex format using a universal reverse primer and four group-specific forward primers. PCR products were analysed by electrophoresis in 2% agarose gels.
Table 1. Primers and conditions used in PCR assays
SCCmec typing
SCCmec PCR typing was based on three separate multiplex-PCR assays and a specific single PCR reaction according to published methods [Reference Zhang17, Reference Ito18]. The first multiplex-PCR assay contained nine pairs of specific primers for SCCmec types and subtypes I, II, III, IVa, IVb, IVc, IVd, and V, and the primers for the mecA gene. The second and third multiplex-PCR assays were used for characterization of mec gene and ccr gene complexes, respectively, and contained four primers each. A single target PCR was used to determine type 5 ccr using ccrC-F and ccrC-R primers. All PCR assays were performed using 100 ng genomic DNA as the template in a PCR mix containing 3·0 mm MgCl2, a 10 mm concentration of the deoxynucleoside triphosphates, the appropriate concentration of each forward and reverse primer, and DreamTaq DNA polymerase under appropriate conditions [Reference Zhang17].
Partial least squares discriminant analysis (PLS-DA)
In order to evaluate if the presence/absence of specific genes correlated with the classification of isolates as HA-MRSA and CA-MRSA, or with the type of clinical sample in HA-MRSA [blood vs. various biological samples (VBS)] two 1/0 matrices were constructed based on results of PCR assays. Logistic PLS-DA, included in SIMCA-P+ software (Umetrics, Sweden), was performed to assess the model's predictability and gene importance in classifying strains. Data were automatically mean centred and unit variance scaled by the statistical software. Each gene was hierarchically classified based on a software-assigned variable importance (VIP) value. The variables with VIP value >1 were chosen as discriminatory for each strain belonging to class (i.e. 1=HA-MRSA and 0=CA-MRSA or 1=blood and 0=VBS). Data were presented as a dendrogram, in which proximity of each gene variable to class centroid (1, 0) indicates the relevance for inclusion in that class.
Evaluation of significance of differences for positivity of PCR assays of single genes were performed by χ2 test at significance levels of P⩽0·05 and P⩽0·01.
RESULTS
Microbiological analyses
Five hundred and seventy-five S. aureus isolates were recovered from 55 057 samples analysed during the study [27499 (49·9%) urine samples, 7152 (13%) blood cultures, and the remaining 20 406 (37·1%) from various sources]. Two hundred and fifty-eight (44·9%) S. aureus isolates were from medical departments, 184 (32%) from surgical departments, and 133 (23·1%) from services in the surrounding area. Of the 575 S. aureus isolates, 211 (36·7%) were from blood cultures and 364 (63·3%) from other specimens. The distribution of S. aureus isolates from blood and VBS from either surgical or medical departments is illustrated in Figure 1. Most blood isolates were from intensive-care wards, although the highest number of isolates in the same sample type was from medical and infectious disease wards.
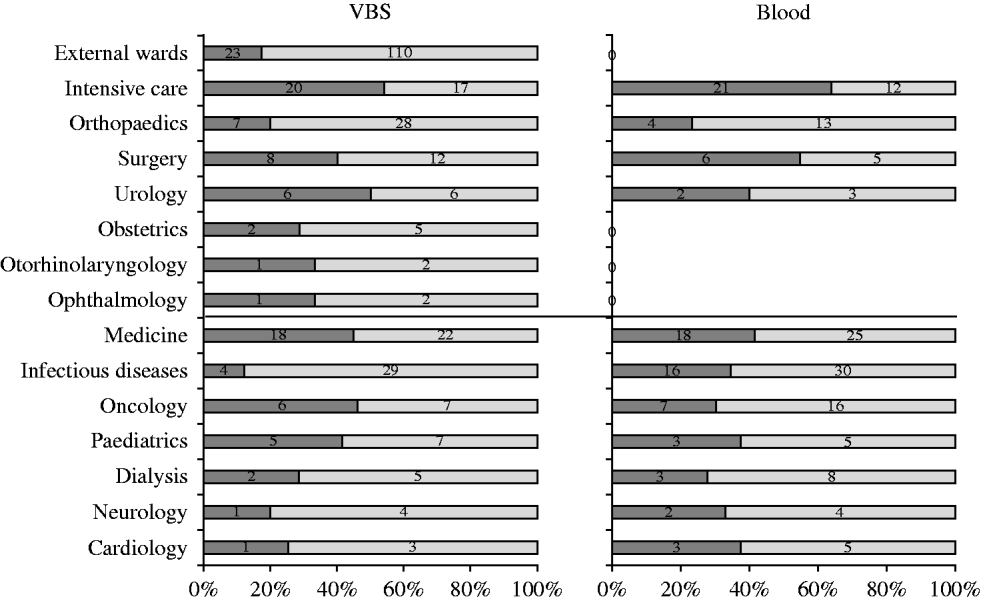
Fig. 1. Distribution of S. aureus isolates (%) in blood samples and various biological samples (VBS) from surgical and medical departments (separated by central black line). ![]() , MRSA.
, MRSA.
Table 2 shows the distribution of methicillin-susceptible (MSSA) and MRSA phenotypes in S. aureus isolates from both blood samples and VBS in HA and CA infections. Of the 575 isolates, 385 (67·0%) were MSSA, and 190 (33·0%) were MRSA. Of the 141 MRSA strains isolated from HA infections, 45 (31·9%) were from surgical, and 63 (44·7%) from medical departments (data not shown). The percentage of MRSA from blood cultures was higher in intensive-care wards (63·6% of 33 isolates) than in surgical wards (54·5% of 11 isolates) and the highest incidence of MRSA in VBS originated from intensive-care wards (54·1% of 37 strains) (Fig. 1).
Table 2. Distribution of the MRSA and MSSA phenotypes in 575 S. aureus isolates from blood samples and various biological samples (VBS) in hospital-acquired and community-acquired infections
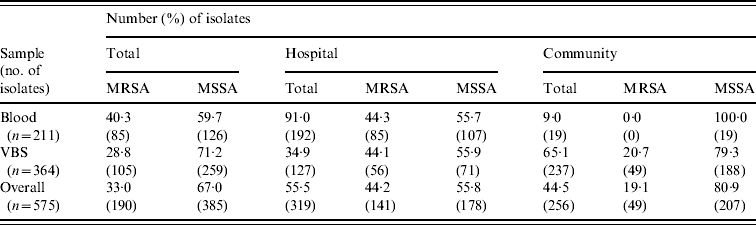
MRSA represented 44·2% of the 319 S. aureus isolates from HA infections. As expected, these isolates were resistant to many of the tested antibiotics (Table 3). By contrast, MRSA represented only 19·1% of the 256 S. aureus isolates recovered from CA infections and these exhibited less antimicrobial resistance compared to HA-MRSA isolates (Table 3). Significant differences (P⩽0·01) were observed in the proportion of resistant strains between MRSA and MSSA for ciprofloxacin, clindamycin, erythromycin and gentamicin in the HA group and for erythromycin in the CA group (Table 3). All MRSA strains were susceptible to linezolid, teicoplanin, and vancomycin (data not shown).
Table 3. Resistance patterns of hospital-acquired (HA) and community-acquired (CA) MRSA and MSSA isolates
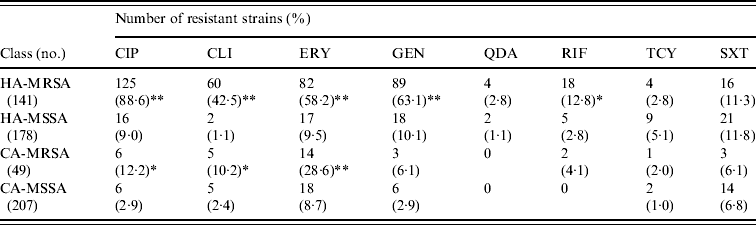
CIP, Ciprofloxacin; CLI, clindamycin; ERY, erythromycin; GEN, gentamicin; QDA, quinopristin/dalfopristin; RIF, rifampicin; TCY, tetracycline; SXT, sulphamethoxazole/ trimethoprim.
* P⩽0·05, ** P⩽0·01 compared to the corresponding MSSA.
agr typing of MRSA strains
Of the 190 MRSA strains isolated during the study, 103 (54·2%) were agrI, 52 (27·4%) agrII, 29 (15·3%) agrIII, and six (3·2%) were agrIV. There were no major differences in the distribution of HA-MRSA and CA-MRSA or origin of sample according to agr type.
Presence of putative virulence determinants in MRSA strains
The distribution of virulence genes in the 190 MRSA strains is reported in Table 4. PCR assays for several genes, including sdrC, sdrD sdrE, clfA, lukS-F PV, and sec, were overall positive in >50% of the strains but cna, seb, and tst were less frequent. Table 4 shows significant differences between HA-MRSA and CA-MRSA isolates for the presence of adhesin (clfA and cna) and toxin genes (etb). There were also significant differences in the gene profiles of isolates from blood and VBS (eap, sec, eta).
Table 4. Frequency of putative virulence genes and SCCmec types detected by PCR in 190 MRSA isolates from hospital-acquired (HA) and community-acquired (CA) infections, and 141 HA-MRSA from blood and various biological samples (VBS). Percentages and number of PCR-positive assays for each gene tested
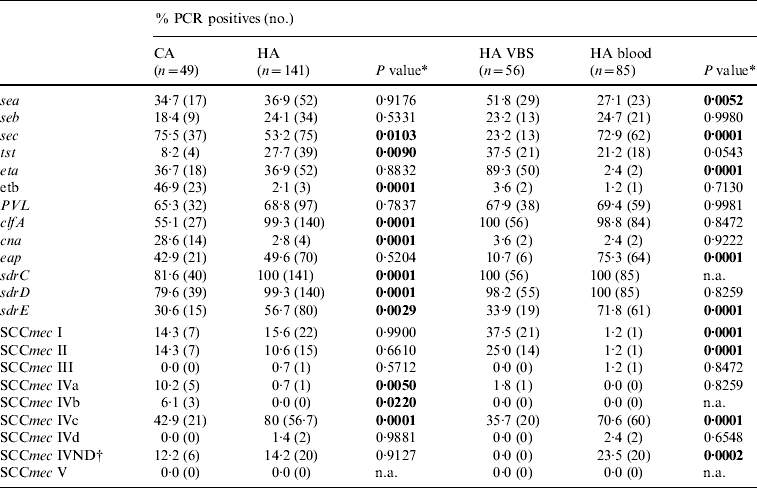
n.a., Not applicable.
* A two-tailed χ2 test with Yates' correction for continuity was applied.
† IV ND: strain showing PCR positivity for more than one type IV SCCmec cassette.
SCCmec typing of MRSA strains
The distribution of SCCmec types in the 190 MRSA strains is reported in Table 4; 138 (72·6%) were SCCmec type IV, with subtype IVc being the most represented (101/190, 53·2%). Twenty of 85 (23·5%) isolates from blood samples harboured SCCmec cassettes that gave specific PCR products for more than one type IV subtype; these strains were classified as not defined type IV (IV ND). Minor, although significant differences in the distribution of SCCmec types between HA-MRSA and CA-MRSA were evident only for the scarcely represented subtypes IVa and IVb (six and three strains respectively). Positivity for subtype IVc and IV ND cassettes was significantly higher in the 85 isolates from blood samples (P<0·01), while types I and II were 21 (37·5%) and 14 (25·0%), respectively, of the 56 isolates from VBS in the HA infections group (Table 4).
PLS-DA analysis
Significant differences in the patterns of selected genes of MRSA isolates classified as HA and CA according to simple temporal criteria, were shown by PLS-DA, resulting in an overall predictability of 93·7%, with a Fisher probability of 1×10−28, and an ANOVA probability of 6·4×10−29 (Fig. 2 a). The PLS-DA model was generated with an overall variance of 71·6% and required the reallocation of 3/141 HA-MRSA (2·13%) as CA-MRSA, and 9/49 CA-MRSA (18·37%) as HA-MRSA. Construction of a dendrogram plotting the hierarchical classification of genes according to variable importance in discriminating HA-MRSA from CA-MRSA showed that genes sdrD and clfA were highly discriminatory for HA-MRSA, and genes cna and etb were highly discriminatory for CA-MRSA (Fig. 2 b).
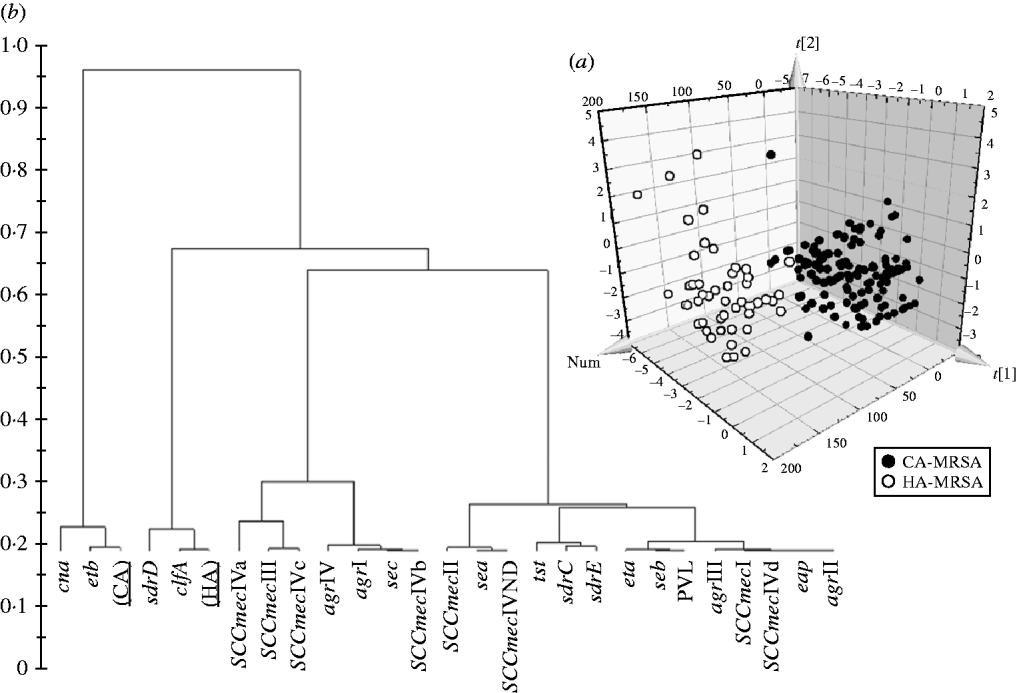
Fig. 2. (a) 3D plot of distribution of the 190 MRSA strains according to presence of specific genes and correlation with the classification of isolates as HA-MRSA and CA-MRSA according to simple temporal criteria, calculated by PLS-DA analysis. (b) Dendrogram of the hierarchical classification of genes for discriminating between groups.
In order to evaluate if the presence/absence of selected genes in HA-MRSA correlated with the clinical source of isolates, the PLS-DA model was performed and gave an overall predictability of 96·4%, with a Fisher probability of 1·1×10−32, and an ANOVA probability of 2·2×10−35 (Fig. 3 a). The PLS-DA model was generated with an overall variance of 82·9% and required the reallocation of 2/56 VBS (3·57%) isolates into the blood class, and 3/85 blood isolates (3·52%) into the VBS class. Construction of a dendrogram plotting the hierarchical classification of genes according to their variable importance in discriminating blood from VBS isolates, showed that genes eta and, to a lesser extent, etb, lukS-F, sdrD and mec IVc cassette, were highly discriminatory for VBS isolates, whilst genes eap, sec, sdrE, and mec IV ND cassette were highly discriminatory for blood isolates (Fig. 3 b).
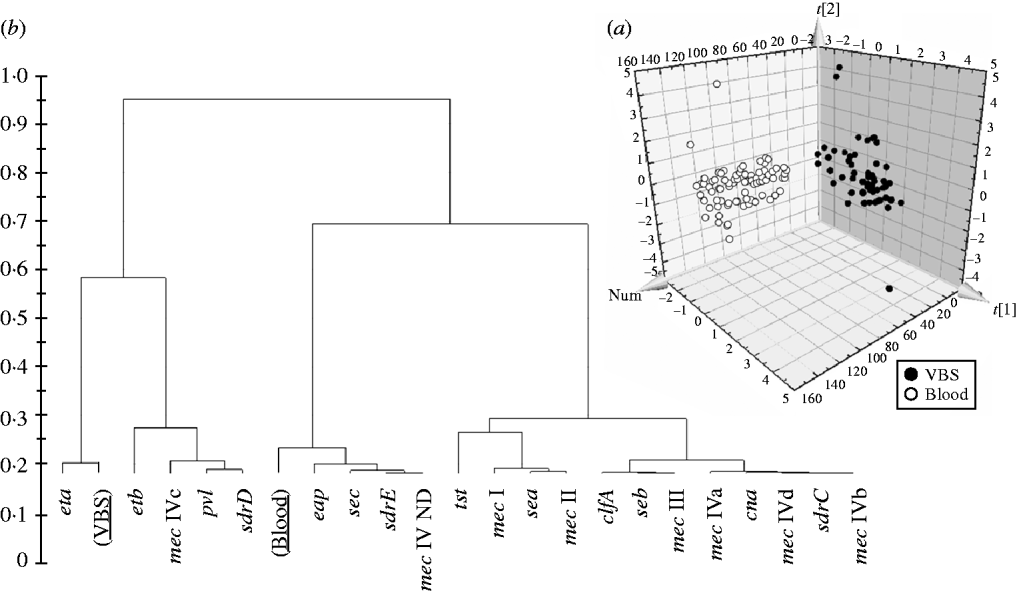
Fig. 3. (a) 3D plot of distribution of the 141 HA-MRSA strains according to presence of specific genes and correlation with the isolation of strains from blood or various biological samples (VBS), calculated by PLS-DA analysis. (b) Dendrogram of the hierarchical classification of genes for discriminating between groups.
DISCUSSION
In this paper we report data on S. aureus isolations of both HA and CA infections in patients attending Viterbo Hospital and the surrounding areas in the period January 2003 to June 2008. During this period S. aureus was isolated from 1·04% of samples tested. Our analysis was particularly focused on MRSA, because during the last few years they have become a great public health concern, due to increasing morbidity and mortality rates and longer hospitalization times. Recent Italian data investigating the incidence of MRSA and characterizing isolates at the molecular level [Reference Stefani and Varaldo5–Reference Campanile7] revealed a constantly evolving situation making it necessary for surveillance to be conducted at both national and local levels. The prevalence of MRSA in the district of Viterbo during the period of this study was aligned to national data.
We defined HA infection as one that developed within 48 h after admission, up to 3 days after discharge, or up to 30 days following surgery. As expected, HA-MRSA were frequently resistant to several antibiotics, while CA-MRSA were generally resistant to β-lactams alone. The PLS-DA analysis evaluating the distribution of specific genes in the isolates confirmed that this simple classification method is reliable and divided them into two distinct and well separated groups, one of which contained all but three of the HA-MRSA and nine strains originally classified as CA-MRSA. Only 5/12 reclassifications (all CA to HA) of the strains showed a prevalence of genetic characteristics of the other group >70%, while the remaining seven strains showed intermediate characteristics. These observations may be indicative of a limited but relevant spread of HA-MRSA into the community, which could prove to be a serious problem by allowing the transfer of resistance genes from HA-MRSA to strains circulating in the community, potentially arming them with increased virulence determinants. This hypothesis is supported by the observation that in a limited but significant number of cases, strains that were originally classified as CA-MRSA, in spite of a further and careful re-evaluation that confirmed classification, were resistant to most of the tested antibiotics. Each of the nine CA-MRSA that were re-classified as HA-MRSA showed atypical resistance patterns; all were resistant to erythromycin and sulphamethoxazole/trimethoprim, and there was variable susceptibility to ciprofloxacin, gentamicin, clindamycin, tetracycline, and rifampicin. Their genetic similarity also correlated with antibiotic resistance patterns as three strains that were genetically most related to HA-MRSA proved to be resistant to all antibiotics tested with the exception of rifampicin susceptibility in a single strain.
SCCmec IVc was more prevalent in HA-MRSA while all other subtypes were more prevalent in CA-MRSA. These data suggest that strains harbouring SCCmec IVc cassettes might host other resistance genes responsible for the multi-resistant phenotype.
Moreover, the distribution of virulence genes was not typical in the MRSA isolates in the study. CA-MRSA are generally regarded as having more virulence genes than HA-MRSA and production of the PVL toxin is normally much more frequent in these strains [Reference Boyle-Vavra and Daum12]. In our sample, the majority of MRSA isolates were PVL positive and were equally distributed in HA-MRSA and CA-MRSA although significant differences were found between the groups for genes encoding adhesins and toxins. Several recent reports suggest that this situation could be a consequence of the strong selective pressure generated by overuse of antibiotics that accelerated the evolution of S. aureus and eventually broke interlineage barriers allowing the frequent acquisition and/or loss of virulence genes carried by mobile genetic elements [Reference Argudìn8, Reference Waldron and Lindsay19]. The widespread diffusion of virulence genes to all agr types in our strain seems to be in accord with this hypothesis.
Our data serve to show the existence of significant genetic diversity between HA-MRSA strains with respect to the site of isolation as a small number of PCR assays could separate with high predictivity isolates from blood and VBS. The non-homogeneous distribution of virulence and resistance genes in isolates from different samples might possibly indicate that interruption of interlineage barriers enables S. aureus strains to rapidly enhance their fitness to different hosts and tissues. Such events could be facilitated by the enhanced circulation of strains of different origin (HA or CA) in the same environments and hosts, as a consequence of public health policy measures directed at reducing costs by shortening periods of hospitalization.
Overall the data presented underlines the fact that the clinical relevance of distinction between HA-MRSA and CA-MRSA is becoming less evident for treatment plans and clinical outcomes. Moreover, the data show that simple criteria that have been developed in the past remain a valid approach for the classification of the origin of an MRSA infection. The findings of our study support the importance of coordinated national and local molecular monitoring of MRSA isolates as a fundamental instrument to inform public health measures designed to reduce the risks associated with the diffusion of infections caused by MRSA both in hospitals and the community.
ACKNOWLEDGEMENTS
This work was supported by a grant from MIUR (Project PRIN 2007LXNYS7_003) granted to C.P.
DECLARATION OF INTEREST
None.







